Shedding light on new treatments for vision, eye problems
The Search Magazine Article | November 15, 2018

The eyes may be the window to the soul, but the laboratory mouse is the window to the eyes and brain.
Research in the mouse eye is shedding light not only on the mechanisms of eye diseases such as glaucoma and macular degeneration, but also Alzheimer’s and other neurodegenerative diseases and even autism. And recent success in delivering corrected genes directly to the mouse eye holds promise for safe and effective gene therapy for inherited eye diseases.
Over the past decade, Jackson Laboratory (JAX) Professor Patsy Nishina and colleagues have developed hundreds of mouse models for translational vision research. Her lab recently announced seven new models, each of which carries genetic variants previously linked to retinal developmental or degenerative ocular disease, and that are now available to the biomedical research community.
In people, most eye diseases take many years, even decades to appear... That's why we need mouse models.
“In people, most eye diseases take many years, even decades to appear,” Nishina says. “This means you don’t know you have the genetic predisposition for the disease until you actually have the symptoms. That’s why we need mouse models: We can develop mice with the same genetic profile as patients, so we can find therapies to target the pre-symptomatic stage to prevent, delay onset or decrease severity of the disease.”
Also, she notes, thanks to the relatively short lifespan of a mouse — about two years in laboratory care — and the ability to control the environmental factors that may contribute to eye diseases, researchers can do detailed, longitudinal studies that reveal how the diseases progress.
In her own research, Nishina focuses on heritable retinal disorders. She and Mark Krebs, Ph.D., an associate research scientist in her lab, are the principal investigators of a grant from the National Eye Institute to study diseases of the retina including retinitis pigmentosa, Leber’s congenital amaurosis and Coat’s disease.
The retina is the structure at the back of the eye that processes visual information. When someone emails you a jpeg, it’s not an image until you open it on your computer. Likewise, the eyes detect light and deliver the information to the brain, which does the job of interpretation.
To get to the retina, light reflects off an object and enters the clear, window-like outer part of the eye, the cornea. Next it travels through the fluid of the anterior chamber to the pupil, the dark circle in the middle of the iris, which is the colored part of the eye that controls how big the pupil is (contracting in the presence of bright light, expanding in dim). Behind the pupil is the lens of the eye, which changes shape when your focus shifts between distant and nearby objects.
Finally, light reaches the retina. It’s made up of millions of photoreceptors — rods and cones — and other cells, packed into a three-layer film only half a millimeter thick over the surface of the back of the eye. These cells convert light into electrical signals that travel along the optic nerve, a bundle of about a million nerve fibers, to the brain.
Nishina shares interest in the retina, as well as lab space on the second floor of the JAX North Research Building, with her husband, Professor Juergen Naggert. He recently received National Eye Institute funding for research in an eye structure known as the external limiting membrane, which could hold the key to understanding and treating enhanced S-cone syndrome (ESCS), diabetic retinopathy and other diseases of the retina.
The adult human retina has about 120 million rods and 6 million cones. Cones come in three subtypes according to the wavelength of light they process: short (S), medium (M) and long (L). In a normal retina the S cones make up just 10 percent of cones, but in ESCS they are the most populous subtype. This causes dysplasia (deformation) of the retina and progressive degeneration of photoreceptors, leading to early night blindness and loss of visual acuity.
Exploiting modifiers
“ESCS is a rare disease caused by mutations in a gene, NR2E3,” says Naggert. “There is quite a bit of variability in the severity of the disease in humans. And in mouse models, the amount of degeneration varies depending on the genetic background of the mouse carrying the mutation. So it’s clear there are ‘modifier’ genes involved.”
Just as every one of us carries genetic variations that predispose us to different diseases, we also have genes that may mitigate the severity of those diseases. But while these so-called modifier genes offer obvious opportunities for treating all kinds of diseases, the complex genetic interactions involved have been extremely difficult to pin down.
This is where special, genetically varied mouse populations come in. Nishina and other JAX eye researchers are using Collaborative Cross and Diversity Outbred (CC/DO) mice to study eye diseases. The product of carefully orchestrated crosses of inbred mouse strains, these mouse populations reflect even more genetic diversity than the entire human population. So it’s possible to trace the progress of a given eye disease in many individuals, and deduce which additional genes may be contributing to more or less severe disease symptoms.

View of the front portion of the eye, including the Schlemm’s canal, blood vessels and lymphatics labeled in yellow and nuclei in blue. Photo courtesy Krish Kizhatil.
The retina: window to the brain
The retina’s interaction with the brain makes it a good and relatively accessible platform for studying neural development. JAX Professor Rob Burgess on a mechanism that allows developing neurons in the mouse retina to connect with one another without excessively clumping together. “A molecule called DSCAM acts like a kind of ‘nonstick’ signal,” Burgess says.
DSCAM and related molecules have also emerged as key contributors to a variety of neurodevelopmental disorders, including autism, schizophrenia, bipolar disease, Down syndrome and intellectual disability. “Indeed, de novo mutations inDSCAM have been linked to autism spectrum disorder in three families,” Burgess notes.
Gareth Howell, a JAX associate professor who studies Alzheimer’s disease (AD), is also the Diana Davis Spencer Foundation Chair for Glaucoma research. It turns out that there are several connections between AD and glaucoma.
In glaucoma, the clear fluid that flows continuously through the anterior chamber at the front of the eye doesn’t drain properly, leading to a buildup of pressure that can damage the optic nerve, leading to vision loss and blindness. An estimated 60 million people worldwide, and 3 million Americans, have glaucoma, and according to the World Health Organization it is the second leading cause of blindness (after cataracts).
Just as a skin infection causes immune cells to rally to the site to mount an inflammatory response to fight it off, nerve cells have their own army of protective immune-like cells, including microglia, and immune cells such as monocytes and macrophages found in the optic nerve head or the brain.
“These immune-like support cells help to clear amyloid plaques in the brain in Alzheimer’s disease and respond to high intraocular pressure in the case of glaucoma,” Howell explains. Harnessing the immune-like response could point to new treatment strategies for both diseases.
breaking-the-bottleneck
Howell and his lab have also identified a protein called MEOX2, which appears to have a role in plaque deposition in AD. “Our gene profiling studies predicted it also played a role in glaucoma,” he says. The team led by Research Intern Rebecca Buchanan and Tufts PhD student Kate Foley looked to the DBA/2J mouse, which develops glaucoma as humans do, in middle age (8-13 months for a mouse). “If you reduce protein levels of MEOX2, glaucoma is worse,” Howell says. “We want to know if there are ways to control MEOX2, or its downstream targets, to switch the environment from a damaging one to a beneficial one.”
“In addition,” Howell says, “we are looking at the eye as a window to the brain for diseases like AD. We’re particularly interested in blood vessel health in the brain. We hypothesize that in AD retinal vessels may decline in a similar fashion to brain vessels. If proven, it would allow us to use retinal vessel health as a proxy for brain vessel health to identify those at risk of AD before dementia-like symptoms develop.”
The mouse and the macula
A leading cause of vision loss in older people is age-related macular degeneration (AMD). According to the National Eye Institute, AMD affects more than 1.75 million Americans, a number that is expected to increase to almost 3 million by 2020 due to the aging of the U.S. population.
Howell is exploring the intriguing interplay between two gene variants,APOE2 and APOE4 that relate to both AMD and AD. The APOE4 variant is the greatest genetic risk factor for late-onset AD, the most common form of the disease. APOE2, on the other hand, appears to have a protective, modifier effect against AD. But in AMD, those roles appear to be reversed:APOE2 raises risk while APOE4 lowers it.
AMD disease causes damage to the macula, a small spot near the center of the retina and part of the eye that is necessary for seeing things clearly. Despite the widespread use of laboratory mouse models in the study of glaucoma and many other eye diseases, research in AMD has been hampered because mice aren’t thought to have a macula.
Krebs in Nishina’s lab has discovered that AMD-like pathology in a recently described mouse model is localized to a small area at the back of the eye, which may be related to the macula.
Working with a mouse model of a macular disease known as butterfly-shaped pigment dystrophy, Krebs observed unusual lesions in a specific area of the retinal pigment epithelium (RPE), a very thin, pigmented cell layer found directly beneath the photoreceptor cells in the retina.
“Our hypothesis is that this area of the mouse posterior eye constitutes a macula-related specialization that develops lesions under conditions associated with human macular disease, including aging,” says Krebs.Nishina and Associate Professor Greg Carter are investigating how disruptions in the RPE can lead to severe vision impairment and loss in AMD and other heritable retinal diseases.
Currently there are no effective cures for AMD or most RPE-related diseases. Previous studies have shown that mouse models of inherited RPE-driven disease often share similar disease features. By examining these models, and using computational tools that the Carter lab is developing, the researchers hope to discover some potential molecular pathways that could be druggable targets.
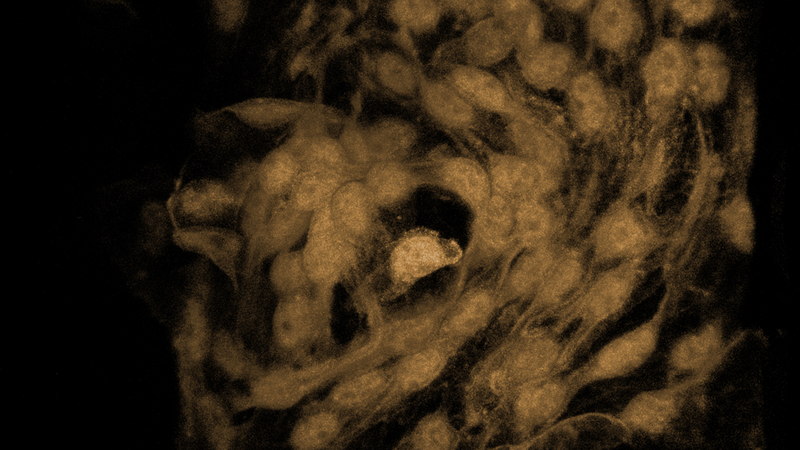
Illustration of Schlemm's canal. Photo courtesy Matt Wimsatt.
New approaches to treatment delivery
JAX Professor and Howard Hughes Medical Institute Investigator Simon John showed that a single, targeted X-ray treatment of an individual eye in young, glaucoma-prone mice provided that eye with apparently lifelong and complete protection from glaucoma. Further research in other animal models to assess protection as well as safety and efficacy is necessary before attempting human treatments, but this work raises the possibility of using highly controlled localized radiation to prevent human glaucoma.
The John lab pioneered the use of laboratory mice for glaucoma studies, and was among the first to show that mice have a structure called Schlemm's canal that is very similar to that in humans., a research scientist in the John lab, focuses on determining the molecular mechanisms functioning in Schlemm’s canal inner wall that facilitates aqueous humor drainage and controls intraocular pressure.
Kizhatil has identified a gene that is a key regulator of lymphatic development and function is expressed in the inner wall of the Schlemm’s canal. This gene appears to control intraocular pressure, and Kizhatil’s studies of this candidate gene could yield critical new information on the mechanisms regulating intraocular pressure, laying the groundwork for identification of more effective therapeutic targets for glaucoma.
Having uncovered a role for L-DOPA in congenital glaucoma, John showed that prenatally feeding the molecule to mice with a mutation in a human developmental glaucoma gene prevents the developmental abnormalities. This suggests that ingestion of foods rich in L-DOPA (such as fava beans) may be therapeutically useful, and may explain why some individuals with congenital glaucoma mutations are protected from disease in populations with a diet rich in L-DOPA.
vitamin-b3-glaucoma-study
Likewise, the John lab more recently showed that in mice genetically predisposed to glaucoma, but who have not yet developed symptoms, Vitamin B3 added to drinking water prevents the disease when administered before symptoms occur, and reduces damage and vision loss in mice that have already developed symptoms. These surprising findings offer promise for developing inexpensive and safe treatments for glaucoma patients via diet or gene therapy, and clinical trials are now in partnership with Columbia University Department of Ophthalmology in New York.
A longtime goal of genetics research has been to develop safe and effective gene therapy: Directly delivering corrected genes to replace defective versions. Significant technical challenges have prevented gene therapy for many diseases, but some of the first successful applications have been in eye diseases, including the John lab’s use of viral vectors to deliver Vitamin B3 therapy to the mouse eye.
In 2007, JAX Research Scientist Bo Chang, director of the JAX Eye Mutant Resource, was on the research team that reported the first successful gene therapy to correct cone cell-mediated electroretinogram (ERG) function and restore sight in mice. Since then Chang has collaborated on several other gene therapy projects, most recently with researchers at the Icahn School of Medicine at Mount Sinai toregenerate rod photoreceptors in damaged mouse retinas.
“Serious or disabling eye diseases affect many millions of people worldwide,” Chang says. “But research on these diseases is limited by the obvious obstacles to studying disease processes in the human eye. Mouse models of inherited ocular disease provide powerful tools for understanding disease progression, and for designing molecules for translational research and gene-based therapy.”