Investigates functional genomics and epigenetic modifications, uncovering how genetic variation and chromatin organization impact development, recombination, and addiction.

Investigates functional genomics and epigenetic modifications, uncovering how genetic variation and chromatin organization impact development, recombination, and addiction.
Genetics is the study of heredity and variation. The Baker Laboratory is ultimately interested in understanding how genetic variation manifests into phenotype. While all of us carry similar genetic code, we recognize and celebrate the fact that small differences provide the building blocks for the wealth of diversity we see among and between populations. It is also this genetic variation that can lead to differences in normal health, susceptibility or resilience to disease, and the origins of complex traits. Research over many years has identified that most of these genetic differences we carry in our DNA are not within genes, but rather in the regions of the genome which regulate how these genes are expressed. Furthermore, these regulatory regions are often transiently active during early development; and we operate under the hypothesis that genetic variation in regulatory activity during early development can have a big impact on adult phenotypes.
Between the genetic code in DNA and functional expression of genes there is a regulatory landscape of chromatin. Chromatin is the physical complex of proteins and DNA; however, chromatin is not static but rather is a dynamic substrate in which gene regulatory elements interact with a host of protein factors to fine tune cell-type specific genome function. Our primary focus is understanding how genetic variation impacts chromatin organization, downstream gene expression, and ultimately cell fate.
Much of genetic research in model organisms relies on a reductionist approach of studying one or a few mutant genes on otherwise isogenic backgrounds. Our goal is to use transformative genomic technologies to embrace genetic complexity and integrate systems genetics into meaningful biomedical research. The Baker Laboratory currently uses three biological systems to understand the effect of genetic diversity on chromatin and cell fate and function:
Mouse embryonic stem cells (mESCs) offer a range of advantages, including scale, flexibility, economy, experimental control, and importantly, a reduction in animal use. Because mESCs are pluripotent, a cellular genetic system enables systematic exploration of gene-environment interactions across large numbers of biomedically relevant cell types, cell states, and organoid cultures. This approach is therefore relevant across a broad range of applications including regenerative medicine.
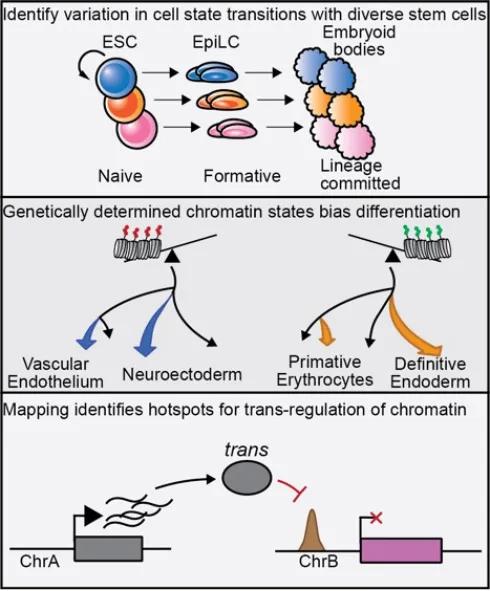
Genetically diverse pluripotent stem cells display varied, heritable responses to differentiation cues. The Baker Laboratory (also in collaboration with the Munger and Reinholdt Laboratories) has recently demonstrated that both local (cis) and, importantly, distal (trans) genetic loci regulate RNA abundance and chromatin accessibility (Skelly et al. 2020, Byers et al. 2021), impacting cell-state transitions. Upon transition to formative pluripotency, some strains quickly dissolved naïve chromatin and gene expression networks and adopted gene expression modules indicative of neuroectoderm lineages; whereas others retained aspects of naïve pluripotency. Spontaneous formation of embryoid bodies identified divergent differentiation wherein ESCs derived from C57BL/6J (B6) mice showed a propensity towards neuroectoderm, while strains such as DBA/2J (D2) tend to differentiate towards definitive endoderm. Genetic mapping of these differences identified majortrans-acting loci co-regulating chromatin accessibility and gene expression in both naïve and formative pluripotency. These loci distally modulated DNA occupancy of pluripotency transcription factors at hundreds of regulatory elements. For example, onetrans-acting locus primarily impacted chromatin accessibility in mESCs; while in epiblast-like cells the same locus subsequently influenced expression of genes enriched for neurogenesis, suggesting early chromatin priming. These results demonstrate that genetically determined variation in chromatin guide certain genotypes towards lineage commitment and allows us to map and identify major regulators of the pluripotency epigenome (Figure 1, reproduced from Byers et al. 2021). Going forward, we are excited to determine how these trans-acting loci influence cell fate and differentiation propensity using techniques in single-cell biology, CRISPR-based screens, and models of developmental disorders.
Meiosis is a specialized cell division responsible both for reducing the number of chromosomes in gametes (i.e., sperm and egg) by half of the normal complement, and for equally partitioning homologous chromosomes between daughter cells. Genetic recombination is the physical breakage and exchange of DNA between homologous chromosomes, and therefore is critical in shaping evolution, increasing genetic diversity, and maintaining genome integrity. Regulation of chromatin therefore plays an essential role in meiosis. In recent years we have learned much about the role of chromatin regulation in determining locations of the double-strand DNA breaks (DSBs) required for genetic recombination; however, key knowledge gaps remain, including an understanding of how chromatin regulation at recombination sites determines the rate of recombination and the ability to repair DNA—essential steps in maintaining genome integrity. The broad goal of this project is to determine the genetic and molecular mechanisms required to establish the permissive chromatin environment for successful recombination and repair.
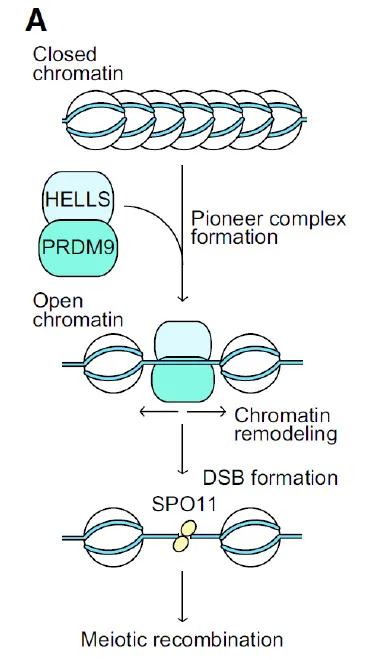
Chromatin-modifying proteins determine the location of recombination. In many mammals, including humans and mice, these locations are determined by the enzyme PRDM9. PRDM9 contains a catalytic domain and an array of tandem C-terminal zinc-finger domains that bind DNA. Following DNA binding, the PRDM9 catalytic domain modifies chromatin with the epigenomic mark histone H3 lysine 4 trimethylation (H3K4me3), often associated with active chromatin. Within populations,PRDM9 is highly polymorphic, with most differences concentrated in the DNA encoding the DNA binding domains. Variability in sequence and number of DNA binding domains allows different PRDM9 protein variants to recognize different DNA sequences, ultimately enabling activation of recombination at diverse locations. Our work and others have found that PRDM9 determines the location of recombination; however, far less is known about what other factors are required to regulate chromatin at sites of recombination. Recently, the Baker Laboratory identified that the protein HELLS (Helicase lymphoid specific), a chromatin remodeling factor, interacts with PRDM9 in germ cells and is required to open chromatin at hotspots, thus allowing for successful recombination and meiotic progression and fertility (Spruce et al. 2020, Figure 2 – reproduced from Alavattam et al. 2020).
Recombination is regulated at two levels: qualitative (genomic location), and quantitative (the frequency with which a particular location undergoes recombination). Recently, the Baker Laboratory demonstrated the existence of a set of loci that regulate the levels of epigenetic modification at hotspots intrans. Using a genetic reference population of recombinant inbred mice initiated from crosses between B6 and D2 mice (BXD recombinant inbred mice), we mapped quantitative trait loci (QTL) that regulate H3K4me3 levels in enriched spermatocytes. In total, this work identified six major QTL that regulate epigenetic marks at hotspots, and subsequently the frequency of double-strand breaks, intrans. The study also showed that, genetically, this trans-acting system acts in a dominant manner to suppress active histone modification in a tissue-specific manner (Baker et al. 2019).
Addiction is a widespread disorder which has a significant socioeconomic impact in the United States. Because we do not fully understand the influence of genetics and epigenetics on predisposition to addition, very few effective pharmacological treatments for addiction exist. Addiction is highly heritable, but our mechanistic understanding of the link between genetic variation, chromatin, and predisposition to addiction is limited. However, it is clear that genetic variation impacts all aspects of addiction and reward seeking behavior and can be modeled in mice. In collaboration with the Chesler Laboratory and The Jackson Laboratory Center for Addiction Biology, we are working to better understand these epigenomic mechanisms in the striatum, a neuronal structure critical for its role in multiple addiction-related behaviors. To investigate genetic loci controlling addiction, our aim is to identify cell-type and strain-specific variation in the chromatin regulatory landscape. To do so we are investigating a panel of genetically-diverse mouse strains that are either susceptible or resistant to reward-seeking behavior using the power of single-cell multi-omics to look at both chromatin and gene expression in the same cell, permitting deeper precision to identify chromatin regulators in individual cell types. This is one of our newest research ventures and we hope to expand our pilot project going forward.