Improving whole genome sequence data analyses
Research Highlight | February 21, 2024

The result — an ability to sequence and resolve diverse human genomes with high accuracy and near 100% completion — is a large step forward, but it has also created new analysis challenges. And since the first human genomes were sequenced, adding additional diversity across global populations remains one of the most important goals of modern genomics.
Working at The Jackson Laboratory, Assistant Professor Christine Beck, Ph.D., and Computational Scientist Peter Audano, Ph.D., are at the forefront of research involving long-read sequence data. They are particularly interested in structural variants (SVs), where segments of the genome are deleted, inserted, duplicated or inverted. Short-read sequencing, in which the genome is broken apart and sequenced in short (often ~250 base pair) segments, fails to detect or characterize many SVs. Long-read technologies have therefore been vital for revealing new insights into how SVs contribute to human diversity and disease. At the same time, as Beck and Audano present in “Small polymorphisms are a source of ancestral bias in structural variant breakpoint placement,” published recently in Genome Research, adjustments to the technical and analytical infrastructure are needed to make the most of long-read sequencing data.
Breakpoint accuracy
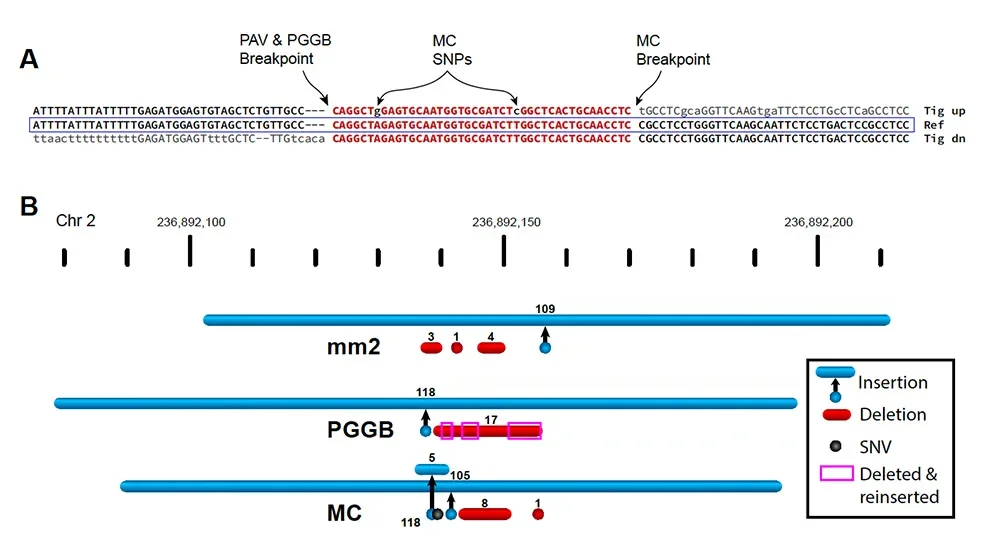
In their analyses of sequence data to investigate the biology of SVs — where they are, how they form, how they affect function and so on — Beck and Audano faced an important problem. While SVs were detected and called out in the analyses, their breakpoints, the exact locations where the standard sequence stops and the altered sequence begins, were identified inconsistently across different samples. They found that a combination of normal variation in the human population and limitations in analysis tools was responsible for different representations of the same SV. Furthermore, these inconsistencies led to an unintentional bias that increases as more diverse populations are analyzed, meaning that valuable information can be lost as a result.
“Three individuals might have inherited the same SV, but current analysis methods might place two of them correctly but the third in a different location based on a nearby single nucleotide polymorphism,” says Beck. “As more and more long-read data is produced, it’s vital for the research community to recognize and address these kinds of systematic ambiguities and errors.”
Work is ongoing to assemble a human “pangenome” that expands and enhances the existing reference genome to include structural and base-pair variation across multiple, genetically diverse individuals. As an early step, adraft pangenome reference was published last May by the Human Pangenome Reference Consortium (HPRC). This adds previously unresolved sequences and more than 1,000 gene duplications to the original reference, as well as roughly 90 million additional base pairs derived from structural variation. The work of Beck and Audano already found additional limitations in pangenome references and thus provides a valuable resource for improving pangenomes. Because populations with greater divergence from the reference are the most impacted by breakpoint location ambiguity and errors, attention is needed to eliminate this bias to support modern genomics and increase the amount of information that is accessible with today’s sequencing technologies.