One gene, two very different diseases
Research Highlight | November 15, 2023
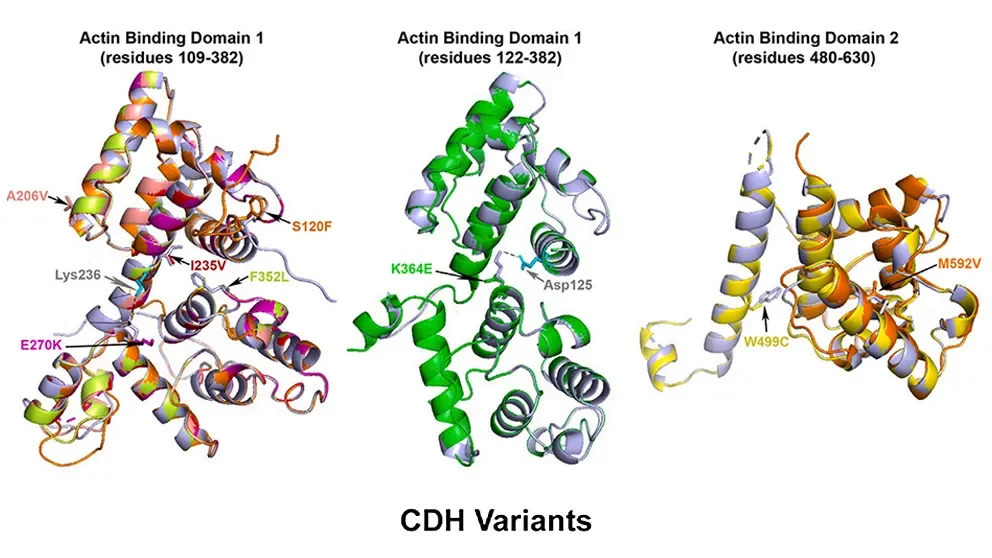

Biomedical research has taught us that simple answers in biology usually tell, at best, only part of a larger story. A recent study led by Frances High, M.D., Ph.D., an assistant professor of pediatrics at Harvard Medical School with appointments at Massachusetts General Hospital and Boston Children’s Hospital, and including Jackson Laboratory ProfessorCarol Bult, Ph.D., is a case in point. The team was investigating the genetics of eight unrelated patients with congenital diaphragmatic hernia (CDH), a relatively common (1 in 3,000 live births) structural birth defect in which diaphragm formation or muscularization is incomplete. Even with advanced medical care and surgical procedures, it is associated with high mortality and morbidity. The researchers found that all eight had what are known as missense variants in plastin 3 (PLS3), a gene that codes for an actin bundling protein involved with cellular structure and movement. But other variants that were predicted to eliminate the same gene’s function had previously been associated with a very different disorder: X-linked osteoporosis in males. So how do different genetic variants in the same gene yield such different conditions?
The effect of CDH on protein function
In “PLS3 missense variants affecting the actin-binding domains cause X-linked congenital diaphragmatic hernia and body-wall defects,” a paper published in The American Journal of Human Genetics, the researchers investigated how the CDH-associated variants might affect protein function. They first modeled the protein structure computationally, which indicated that the resulting proteins probably wouldn’t have major changes in shape or conformation. Nonetheless, because the variants were all located within the gene sequences that code for the actin binding domains, the protein would likely have altered actin binding.
The research team was then able to engineer a mouse model with the exact missense variant found in one of the human CDH patients. The mice exhibited diaphragm abnormalities and body-wall defects, and most died soon after birth. Interestingly, the mice that survived had increased bone mineral density (BMD), while aPls3 loss-of-function mouse model showed lower BMD, mirroring the variant effects seen in human patients.
The CDH-associated variants, which produce what the authors propose calling “PLS3 congenital anomaly syndrome,” are theorized to affect actin cell structure dynamics and, in turn, proper diaphragm and body wall development. They may also have a gain-of-function effect as seen in bone, leading to the increased BMD. In contrast, variants that lead to loss of normal protein function appear to have no effect on diaphragm development but do lead to altered calcium responsiveness and osteoporosis. The very different consequences of the distinct types of genetic variants have clinical implications, including the potential for developing therapeutic strategies for these and related structural anomalies and bone diseases.