Insulin replacement therapy is the primary treatment for type I and advanced type II diabetes. Replacement is required due to the progressive loss of insulin producing pancreatic b-cells in both forms of the disease. Not surprisingly, significant research is being conducted to identify methods to effectively replace the b-cells and restore glycemic control. Pancreatic b-cell function is regulated in part by an incretin hormone, glucagon-like peptide-1 (GLP-1), that is produced by intestinal L cells. After a meal, GLP-1 secretion is stimulated by nutrients in the gut. Once in the circulation, GLP-1 binds to its receptor, GLP-1R, which is expressed by b-cells, and causes insulin release through an adenylyl cyclase mediated pathway. GLP-1 also prevents apoptosis and stimulates both b-cell proliferation and differentiation. Stimulation of another b-cell receptor, G-protein-coupled receptor 119 (GPR119), also increases GLP-1 release and subsequent insulin secretion. Researchers at the University of South Dakota’s Sanford School of Medicine (USD) and the University of Pennsylvania (UPenn) report in the Journal of Diabetes Research (Ansarullah et al 2016) that a small molecule GPR119 agonist increases proliferation of transplanted human b-cells and more rapidly restores normoglycemia in a streptozotocin (STZ)-induced mouse model of hyperglycemia.
GPR119 stimulation in human b-cells accelerates glycemic control in vivo
Prior research demonstrated that a small molecule agonist of GPR119, PSN632408, could stimulate replication and differentiation of mouse b-cells. In their 2016 report, the USD and UPenn teams demonstrated that the same activity can be achieved with human cells in an in vivo setting. The question was addressed using the non-diabetic NOD-scid immunodeficient mouse strain, NOD.CB17-Prkdcscid/J (001303). In their study, NOD-scid mice were treated with STZ to ablate their pancreatic b-cells and induce hyperglycemia. The mice then received human b-cell transplants that were placed under the kidney capsule. One group of mice was treated with PSN632408 (the GPR119 agonist), and the second was treated with vehicle alone. Both groups were monitored for fed blood glucose (mg/dl) every few days for 4 weeks. Non-transplanted mice had diabetic fed blood glucose values averaging ~540 mg/dl. As shown in the figures below, the PSN632408-treated mice showed more rapid return to normoglycemia (A) (that is, glucose values at or below 200 mg/dl). The agonist treated mice had a higher percentage of mice with normoglycemia by 4 weeks post-transplant. At 2 weeks post-transplantation, 38% of agonist-treated mice had normal glucose values versus 0% in the control group (not shown). At 4 weeks, 71% of the treated mice had normal values and only 24% were normoglycemic in the controls (B).
A. 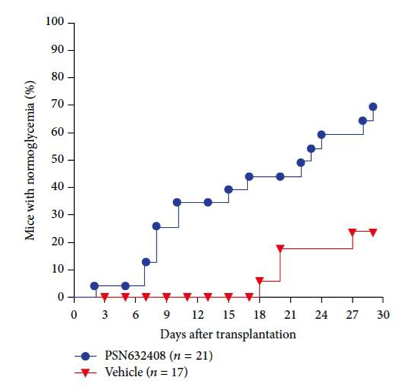 |
B. 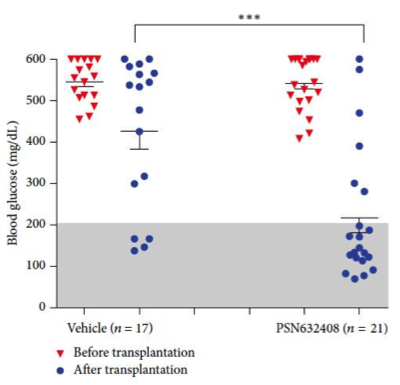 |
| Immunodeficient NOD-scid mice were treated with STZ to deplete mouse β-cells and induce hyperglycemia. Hyperglycemic mice were transplanted with human β-cells to restore insulin production and regain regulation of glucose metabolism. (A) Mice treated with PSN632408 regained control of hyperglycemia at a faster rate and in a higher percentage of mice than in vehicle treated controls by 30 days post β-cell transplant. (B) Four weeks post β-cell transplant, only 4 of 17 (24%) vehicle treated mice had normal fed blood glucose versus 15 out of 21 (71%) PSN632408-treated mice. | |
Human b-cell function in the transplanted mice was tested by glucose tolerance test (GTT). Here, the two groups of mice were fasted and then challenged with an oral glucose bolus, after which the rate at which they cleared glucose from blood was measured. The GPR119 agonist-treated mice cleared glucose more rapidly compared to the vehicle-treated controls, indicating improved b-cell function. To demonstrate that glycemic control was mediated by the transplanted human b-cells and not by residual mouse b-cells, the kidneys containing the human b-cell transplants were removed. Following nephrectomy all mice developed hyperglycemia, showing that residual mouse b-cells were not sufficient to regulate blood glucose. Taken together, these results show that the GPR119 agonist improved glycemic control by the human transplanted b-cells in vivo.
GRP119 agonist induces human b-cells proliferation and expansion.
Examination of the human transplanted b-cells for insulin expression by immunohistochemistry showed that the agonist-treated mice had significantly increased b-cell area compared to controls. In order to determine if the increased b-cell area was due to cell proliferation, cells were incubated with bromodeoxyuridine (BrdU). Proliferating cells will incorporate this synthetic thymidine analog into their DNA and can be detected subsequently by BrdU-specific antibodies. Mice treated with PSN632408 had significantly greater numbers of insulin and BrdU double-positive cells. Kidney transplants also were co-stained for insulin and the proliferation marker Ki67. Here again, the PSN632408-treated mice had a higher percentage of proliferating insulin-producing human b-cells. These data clearly demonstrate that the GPR119 agonist stimulated the human b-cells to proliferate and expand.
GRP119 agonist stimulates human b- and a-cell neogenesis.
Next, the researchers investigated if PSN632408 stimulated neogenesis of new human b-cells. The transplanted human islets contained a mixture of cells, including pancreatic duct acinar cells. Lineage tracing studies performed by other research groups indicated that duct cells are capable of differentiating into b-cells. The pancreatic duct cells can be identified by the cytokeratin marker CK19. Immunohistochemistry revealed that the mice treated with PSN632408 had a higher percentage of insulin-expressing, CK19-positive cells compared to the vehicle-treated controls (3.7% versus 1.3%, respectively).
Other cells found in pancreatic islets that are important in glucose metabolism are a-cells. The a-cells produce glucagon, which induces gluconeogenesis in the liver, and is believed to be involved in b-cell differentiation. Accordingly, the USD and UPenn investigators examined the transplanted human tissues for changes in both human a-cell area and numbers. In agonist-treated mice, 17.5% of the cells in the transplant were glucagon-positive a-cells, versus 10.6% in the controls, indicating an increase in a-cells. A higher percentage of the human glucagon-positive a-cells in the agonist-treated mice were also BrdU positive compared to controls. These results demonstrate that the GPR119 agonist also stimulated a-cell proliferation and differentiation. Therefore, increased a-cells and subsequent production of glucagon may also contribute to b-cell neogenesis in the human tissue transplants following stimulation through GPR119.
The report by Ansarullah et al. identifies an important receptor pathway that can induce the expansion of transplanted human b-cells in vivo. Typically, a diabetic human patient already has reduced numbers of insulin-producing b-cells when they are diagnosed. A treatment strategy to produce more of these cells through neogenesis could reduce the need for insulin replacement therapy. Also, human donor b-cells are in limited supply, but in vivo expansion of transplanted cells could accelerate and maintain improved control of glucose metabolism. Both strategies, however, will need to be coupled with additional treatments. In the case of autoimmune type 1 diabetics, patients also will require a treatment to prevent autoimmune destruction of either host-expanded or transplanted cells. Type 2 diabetics will need to receive a companion treatment that will modify metabolism to allow both decreased glucose production and improved glucose utilization. Regardless, methods to enhance and increase neogenesis of insulin-producing b-cells likely will be important components to new treatment strategies for human patients reliant on life-long insulin replacement therapy.
Reference
Ansarullah, et al., 2016. Activation of GPR119 Stimulates Human b-Cell Replication and Neogenesis in Humanized Mice with Functional Human Islets. Journal of Diabetes Research. Volume 2016 (2016), Article ID 1620821, 12 pages.
Permissions
http://www.hindawi.com/journals/jdr/2016/1620821/
This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.