Ice water fires up ALS research
Recently, the ALS Association announced that it had raised more than $115 million via its ALS Ice Bucket Challenge, a fund-raising effort in which celebrities, athletes, politicians and every-day citizens poured buckets of ice water over their heads and challenged their friends and colleagues to do the same – to raise money for and to increase awareness of amyotrophic lateral sclerosis (ALS) disease research. Although ice bucket challenges have been used as fund-raising vehicles for other charities and disease foundations in the last few years, the ALS Ice Bucket Challenge has been particularly successful due to the rash of videos that went “viral” on social media. For a while, it seemed that every YouTube channel you visited had someone taking the challenge.
ALS is commonly called “Lou Gehrig’s Disease” after the famed New York Yankee first baseman who suffered from the disease. Gehrig first brought ALS to the nation’s attention when he was forced to retire from baseball when the disease began to affect his playing ability. It is a debilitating and progressive motor neuron degenerative disease that manifests itself initially as muscle weakness but progresses rapidly through decreasing motor coordination to general paralysis. Most patients succumb just 3-4 years after diagnosis due to respiratory failure.
In light of the attention that the ALS Ice Bucket Challenge has brought to ALS disease research, it seems timely to review the state of the ALS-related mouse models that are available from JAX and the considerations for using them in research.
Mouse models for ALS: SOD1 and TARDBP transgenic mice
JAX distributes two types of ALS models: mice that overexpress mutant forms of the human copper/zinc superoxide dismutase 1 (SOD1) gene, and mice that overexpress mutant forms of TAR DNA binding protein (TARDBP or TDP-43).
SOD1 transgenic mice
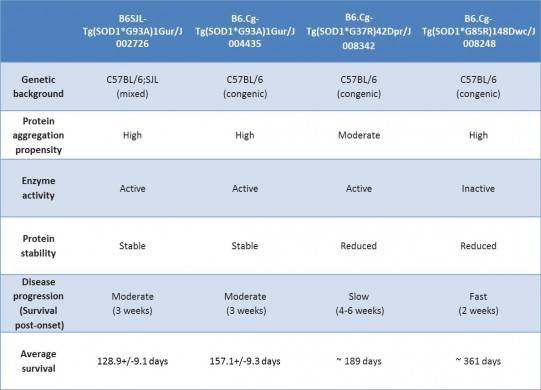
Only approximately 10% of ALS cases show linkage to family-inherited mutations; the majority develop spontaneously. Of the heritable mutations that have been linked to ALS, mutations in SOD1 are the most common, comprising roughly 20% of this group. SOD1 encodes a mitochondrial protein that protects cells from damage from superoxide free radicals produced during normal metabolism. Ubiquitin-positive protein aggregates in motor neurons from patients who carry ALS-linked SOD1 mutations suggests that changes in protein stability, degradation or trafficking may contribute to either mitochondrial dysfunction or cellular damage that may result in neural degeneration. Because motor neurons in Sod1 knockout mice do not degenerate, the ALS-linked SOD1 mutations are likely gain-of-function alleles.
JAX distributes several transgenic mice that overexpress various mutant forms of human SOD1. All of them display many of the histopathological phenotypes that are observed in humans with ALS including accumulation of SOD1/ubiquitin protein aggregates, reactive astroglia and microglia, and motor neuron degeneration, but disease onset and progression vary considerably (see below).
Table 1. Characteristics of featured JAX SOD1 transgenic mice
TARDBP transgenic mice
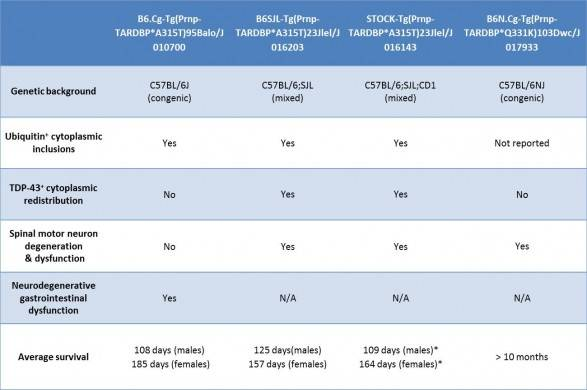
TAR DNA binding protein (TARDBP) is another gene that has been implicated in ALS. TARDPB protein is a major component of the ubiquitin-positive cytoplasmic inclusions that are present in the majority of non-familial ALS patients, and mutations in TARDBP are linked with some familial ALS patients. Genetic deletion of TARDBP results in embryonic lethality, so as with SOD1 mutations, TARDPB mutations in familial ALS patients are likely gain-of-function alleles.
In general, TARDBP transgenic mice develop a neurodegenerative phenotype reminiscent of ALS, but in only some models has redistribution of TARDBP to the cytoplasm been observed. Further, even in mice where increased TARDBP cytoplasmic levels are reported, association of TARDBP with ubiquitin-positive protein aggregates has not been definitively demonstrated. The phenotypes of selected JAX TARDBP transgenic mice are summarized below:
Table 2. Characteristics of featured JAX TARDBP transgenic mice
ALS mouse model phenotypes: It’s more than just the transgene
In both SOD1 and TARDPB transgenic mice, genetic background can significantly influences a strain’s disease onset and severity. With SOD1*G93A transgenic mice, backcrossing to C57BL/6 delayed the onset of disease compared to mice on a C57BL/6 x SJL mixed background. More strikingly, backcrossing Tg(Prnp-TARDBP*A315T)95Balo transgenic mice to C57BL/6J produced mice with an entirely new phenotype that was not reported for the original strain, whose genetic background was a C57BL/6xCBA mixture: on the B6 background death results from a neurodegenerative phenotype in the neurons that innervate the gut in the absence of any motor neuron degenerative pathology.
The sex of the mice, too, can profoundly influence disease outcomes and endpoints in these ALS models.
Tips for working with ALS mouse models
If you are currently working with or planning to use any of these ALS mouse models, check out Working with amyotrophic lateral sclerosis (ALS) mouse models. This resource is a practical manual with guidelines and recommendations for conducting preclinical studies including model-specific tips on how to manage ALS colonies and study design considerations when using mouse models of ALS. Although the guide focuses on the SOD1 transgenic mouse models exclusively, the considerations discussed in the guide are valuable for designing studies using TDP-43 transgenic mice, too.
The ALS Ice Bucket Challenge has been an overwhelming and unexpected success. We hope that some of the funds raised by the initiative will find their way to researchers developing new ALS mouse models and using them in preclinical trials to uncover new therapeutic candidates.