In 2001, the International Human Genome Sequencing Consortium announced the first draft of the human genome reference sequence. The Human Genome Project, as it was called, had taken more than eleven years of work and involved more than 1,000 scientists from 40 countries. This reference, however, did not represent a single individual but instead is a composite of humans that could not accurately capture the complexity of human genetic variation.
Building on this, scientists have carried out many sequencing projects over the last 20 years to identify and catalog genetic differences between an individual and the reference genome. Those differences usually focused on small single base changes and missed larger genetic alterations. The larger differences are called structural variants (SVs) and include DNA segments that are inserted, duplicated, inverted or deleted within the genome. Structural variants are more difficult to detect than single base changes, but they are more likely to interfere with gene function.
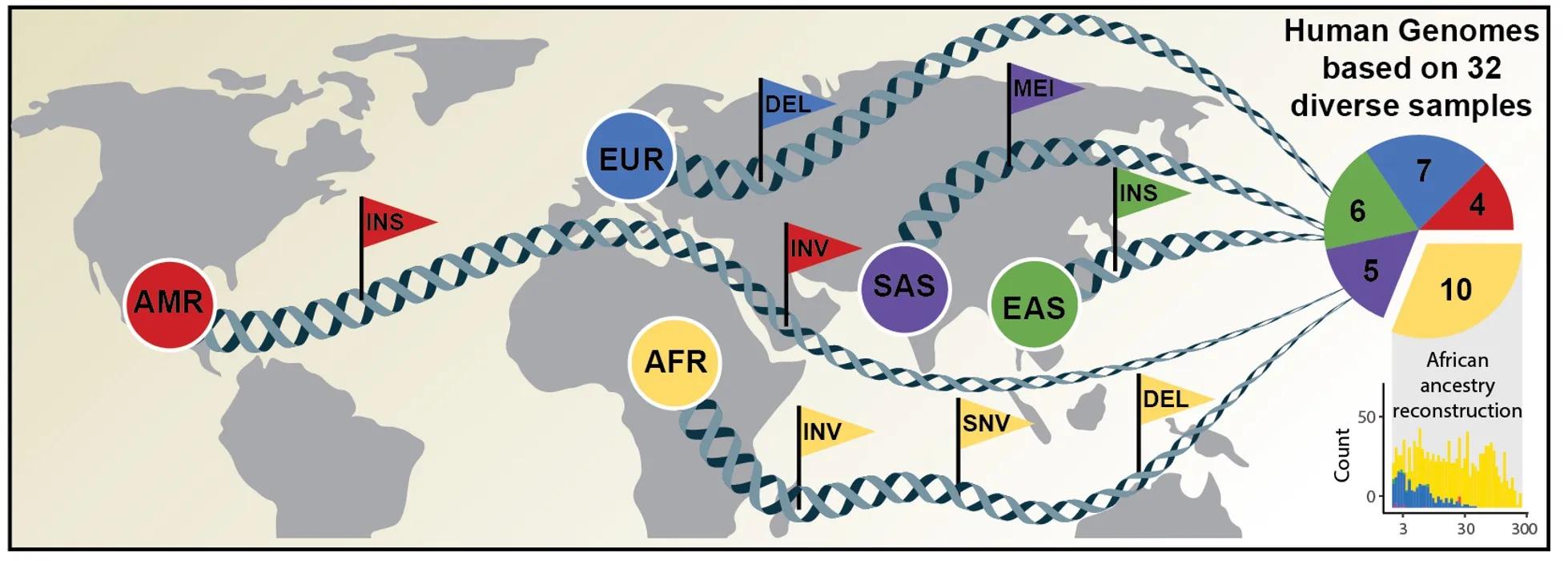
An international research team has now published an article in Science announcing a new, considerably more comprehensive reference dataset obtained using a combination of advanced sequencing and mapping technologies. The new reference dataset reflects 64 assembled human genomes from 32 individuals representing 25 different human populations from across the globe. Importantly, each of the genomes was assembled without guidance from the human genome reference sequence and as a result better captures genetic differences between different human populations. The study was led by scientists from The Jackson Laboratory for Genomic Medicine in Farmington, Conn. (JAX), the University of Washington in Seattle (UW), the European Molecular Biology Laboratory Heidelberg (EMBL), and the Heinrich Heine University Düsseldorf (HHU).
"The first human genome sequence was a huge step forward, but was incomplete," says co-senior author Charles Lee, Ph.D., FACMG, director and professor, The Jackson Laboratory for Genomic Medicine. "In addition to single base variation, we now know that structural variants also contribute very substantially to genomic differences between individuals. Our work provides a far more thorough and accurate window into that genomic variation across individuals and populations, and it represents an incredibly valuable new resource for the research community."
The new reference data now represent the full range of different genetic variant types and incorporates human genomes of great diversity. The aim is to estimate the individual risk of developing certain diseases such as cancer and to understand the underlying molecular mechanisms. This, in turn, can be used as a basis for more targeted therapies and preventative medicine.
"Capturing the full spectrum of structural variation found in human genomes is vital for clinical applications," says Qihui Zhu, Ph.D., computational scientist and co-first author on the study. "These variants affect gene function and can contribute to diseases, drug response differences, and more. Knowing how they differ across individuals and across populations is needed to implement more effective genomic medicine."
This study builds on a new method published by these researchers last year inNature Biotechnology [Link: https://www.nature.com/articles/s41587-020-0719-5] to accurately reconstruct the two components of a person's genome – one inherited from a person’s father, one from a person’s mother. When assembling a person’s genome, this method eliminates the potential biases that could result from comparisons with an imperfect reference genome.
Source
Peter Ebert*, Peter A. Audano*, Qihui Zhu*, Bernardo Rodriguez-Martin*, David Porubsky, Marc Jan Bonder, Arvis Sulovari, Jana Ebler, Weichen Zhou, Rebecca Serra Mari, Feyza Yilmaz, Xuefang Zhao, PingHsun Hsieh, Joyce Lee, Sushant Kumar, Jiadong Lin, Tobias Rausch, Yu Chen, Jingwen Ren, Martin Santamarina, Wolfram Höps, Hufsah Ashraf, Nelson T. Chuang, Xiaofei Yang, Katherine M. Munson, Alexandra P. Lewis, Susan Fairley, Luke J. Tallon, Wayne E. Clarke, Anna O. Basile, Marta Byrska-Bishop, André Corvelo, Uday S. Evani, Tsung-Yu Lu, Mark J.P. Chaisson, Junjie Chen, Chong Li, Harrison Brand, Aaron M. Wenger, Maryam Ghareghani, William T. Harvey, Benjamin Raeder, Patrick Hasenfeld, Allison A. Regier, Haley J. Abel, Ira M. Hall, Paul Flicek, Oliver Stegle, Mark B. Gerstein, Jose M.C. Tubio, Zepeng Mu, Yang I. Li, Xinghua Shi, Alex R. Hastie, Kai Ye, Zechen Chong, Ashley D. Sanders, Michael C. Zody, Michael E. Talkowski, Ryan E. Mills, Scott E. Devine, Charles Lee#, Jan O. Korbel#, Tobias Marschall#, Evan E. Eichler#,Haplotype-resolved diverse human genomes and integrated analysis of structural variation, Science 2021
*Co-first authors #Co-senior and co-corresponding authors
About The Jackson Laboratory
The Jackson Laboratory is an independent, nonprofit biomedical research institution with more than 2,400 employees. Headquartered in Bar Harbor, Maine, it has a National Cancer Institute-designated Cancer Center, a genomic medicine institute in Farmington, Conn., and facilities in Ellsworth and Augusta, Maine, in Sacramento, Calif., and in Beijing and Shanghai, China. Its mission is to discover precise genomic solutions for disease and empower the global biomedical community in the shared quest to improve human health.