Treating melanoma by inhibiting BRAF & PD-L1, coupled with enhanced dendritic cell activity
June 8, 2016
Single-drug cancer therapies are now available that afford some patients long-term and durable tumor remission. Unfortunately, many patients still either relapse, or are non-responders. Researchers and clinicians, therefore, are turning to combinatorial treatments that both kill tumors directly and stimulate the patient’s immune system to overcome tumor-mediated immune resistance. New research reported in the JournalImmunity (Salmon et al, 2016) elegantly teases apart the key cellular players in immune-mediated tumor killing and demonstrated treatment combinations to enhance both the presence and activation of these cells to improve their tumor-killing activities in mouse models for melanoma.
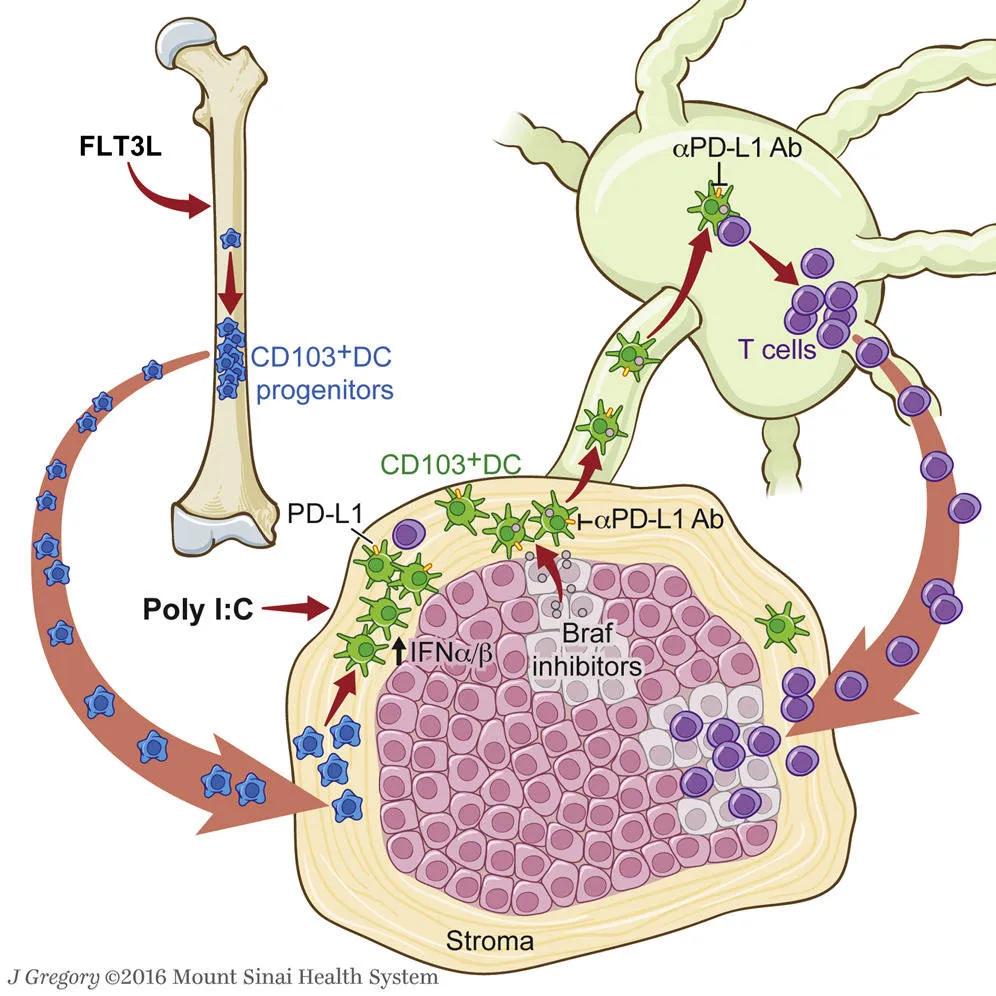
Figure. CD103+ DC are key to melanoma tumor antigen presentation and activation of T cells.
DC are key to melanoma tumor antigen presentation and activation of T cells.
Melanoma tumor resistance to immune- mediated killing can be reversed by a combinatorial therapy involving: (1) FLT3L-mediated increase of the CD103+ dendritic cells (DCs) population infiltrating the tumor; (2) activation of CD103+ DCs to enhance priming of T cells by poly I:C; (3) BRAFV600E inhibitor treatment that decreases tumor cell proliferation and enhances tumor cell death , allowing antigen release to CD103+ PD-L1+ DC, and (4) anti-PD-L1 mAb mediated checkpoint inhibitor blockade.
Tumor infiltrating CD103+ dendritic cells prime T cells with tumor antigens
One approach to treating human patients with melanoma is though inhibiting the MAP kinase proliferation pathway that has escaped normal control due to theBRAFV600E mutation. This mutation is commonly associated with melanoma and many other cancers. Small molecule inhibitors of BRAFV600E decrease proliferation and stimulate cell death without affecting cell that express normal BRAF. Another breakthrough in melanoma treatment is a class of antibodies the bind either PD-1 on T cells or its ligand, PD-L1, which is found on both tumor cells and antigen presenting cells. When T cells engage antigen presenting molecules through their T cell receptor (TCR), they are activated to become either cytotoxic, if they receive additional signals through costimulatory molecules, or anergic if they receive signals from checkpoint blockade pathways, such as PD-1 binding to PD-L1. Antibodies that bind to either of these proteins in a manner that prevents PD-1:PD-L1 engagement, block the anergic signaling pathway and re-animate T cell activation. Not all patients respond equally to these therapies, however; some are resistant and others relapse over time.
Understanding the cells and mechanisms involved in therapeutic response and resistance are the key to developing more durable treatments. In theirImmunity study, the Salmon et al. team used two syngeneic mouse models to approach the problem: first, the B16 melanoma cancer cell line engrafted subcutaneously into C57BL/6 recipients; and second, genetically-engineered and inducible mouse models. Two different inducible models were used: Tyr::CreER, BrafCA, PtenloxP(B6.Cg-Braftm1Mmcm Ptentm1Hwu Tg(Tyr-cre/ERT2)13Bos/BosJ, 013590) and Tyr::CreER, BrafCA, PtenloxP, Ctnnb1loxex3mice. These strains both carry a transgene with a tyrosinase (Tyr) promoter driving tamoxifen-induciblecre recombinase (cre) in cutaneous melanocytes. The BrafCA (Braftm1Mmcm) allele contains a loxP-flanked sequence that when recombined by cre initiates expression of theBrafV600E mutant protein. The PtenloxP (Ptentm1Hwu) allele also is cre conditional, but in this case, cre recombination knocks out expression of this tumor suppressor gene. Finally, in theCtnnb1loxex3 (Ctnnb1tm1Mnmt) allele, exon 3 is flanked by loxP sites. When this exon is removed by cre, Ctnnb1 protein stability is enhanced. Ctnnb1 overexpression disrupts cell gene expression more broadly, and is associated with multiple cancers. In both of these engineered models, topically applying 4-hydroxytamoxifen (4-HT) to the skin allows migration of cre from the melanocyte cytoplasm into the nucleus, where it recombines the loxP-flanked target genes and drives subsequent melanoma development. For simplicity’s sake, these mice will be referred to collectively as 4-HT-induced melanoma models below.
In both the cell line and inducible cancer models described above, the Soloman team characterized the types of immune cells that infiltrated their respective tumors. The tumors contained CD4+ and CD8+ T cells, NK cells, but very few B cells. Late-stage tumors had high numbers of monocytes and macrophages, but very few CD103+ or CD11b+ dendritic cells (DCs). The frequency of myeloid cells in these tumors closely resembled those observed in human myelomas. Myeloid cells are equipped to phagocytize proteins and present antigens, so the researchers interrogated tumor draining lymph nodes (TdLN) to determine which cell types were migrating to the LN and priming T cells. They did this using yellow fluorescent protein (YFP)-transduced B16 tumor cells and by crossing Tyr::CreER, BrafCA, PtenloxPmice to strain B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J, 007914. The latter mice are a cre reporter that expresses red fluorescence following cre recombination. These two models allowed tracking specific myeloid cells that had phagocytized fluorescent protein from the tumor cells and migrated to the TdLN. CD103+ DCs were the only myeloid cell population in the TdLN that was positive for fluorescent protein in both tumor models. Two additional experiments were performed to validate the role of these cells. In the first experiment, YFP-labeled B16 cells were engrafted in chemokine receptor 7 (Ccr7)-deficient hosts (B6.129P2(C)-Ccr7tm1Rfor/J, 006621 ). Ccr7 is a chemokine receptor that is required for DCs to migrate to the lymph node. The TdLN in the Ccr7-deficient mice had very few YFP+ CD103+ DCs compared to hosts with normal Ccr7. In the second experiment, B16 cells modified to express ovalbumin (OVA) were engrafted into normal mice, the recipients’ TdLNs were collected, and DC populations were isolated by flow activated cell sorting (FACS). The FACS-isolated DCs were then cultured with T cells collected from OT-I TCR transgene mice (C57BL/6-Tg(TcraTcrb)1100Mjb/J,003831) , which specifically and almost exclusively express an ovalbumin-specific TRC in CD8+ T cells. Following co-culture, only the CD103+ DCs were capable of activating OT-I-derived T cell proliferation. Collectively these data demonstrate that CD103+ DCs infiltrate tumors, process antigen, migrate to the TdLN, and prime CD8+ T cells with tumor antigens.
FLT3L and poly I:C increase numbers, maturation and activity of CD103+ DC
Once the key tumor antigen presenting cell population was identified, the next questions were:
- Why don’t these cells stimulate a stronger anti-tumor cytotoxic response?
- How can the response be amplified?
Further phenotyping of the CD103+ DCs from TdLNs revealed that these cells express high levels of PD-L1, indicating they may play a role in checkpoint inhibition of PD-1+ T cells. This was investigated by transplanting B16 tumor cells (that also express PD-L1) into both normal, control mice and in mice deficient in Batf3 (B6.129S(C)-Batf3tm1Kmm/J, 013755). Batf3 is necessary for CD103+ DCs development. When mice were treated with anti-PD-L1 monoclonal antibody (mAb,) the normal controls showed diminished tumor growth, indicating activation of cytotoxic T cells towards the tumor. This effect was lost in the Batf3-deficient mice, clearly demonstrating that CD103+ DCs play an important role in both antigen presentation and checkpoint inhibition of T cells.
Although checkpoint inhibitor blockade reduced tumor growth in both the B16 and 4-HT-inducible myeloma models, tumor growth was not completely inhibited. The authors hypothesized that increasing the numbers of activated CD103+ DCs in the tumor might enhance the response. Accordingly, they treated tumor-bearing mice with daily injections of the growth factor FMS-like tyrosine kinase 3 ligand (FLT3L). The FLT3L treatment stimulated MHC II+, CD11c+, CD103-, CD11b-, IRF8+ progenitors in the bone marrow to greatly expand CD103+ DCs in the tumors. The DCs, however, must be activated and functionally mature in order to maximally stimulate cytotoxicity in T cells. CD103+ DCs express toll-like receptor 3 (TLR3) that when bound to double-stranded RNA (dsRNA), stimulates cell maturation and interferon type I (IFN1) production. Type I IFN is required for efficient T cell priming. TLR3 also can be activated using the synthetic dsRNA analog polyriboinosinic:polyribocytidylic acid (poly I:C). Therefore, tumor bearing mice from both melanoma models were treated with FLT3L, poly I:C, or both together (FLT3L + poly I:C). As expected, the mice treated with FLT3L + poly I:C showed the most significantly reduced tumor growth in both models. When the experiment was repeated using the B16 cells in Batf3-deficient mice that lack CD103+ DCs, the effect of FLT3L + poly I:C was lost. Altogether, these results further demonstrated the role of CD103+ DC in the priming of T cells with tumor antigens and that strategies for increasing the numbers of activated tumor-infiltrating DCs could enhance immune-mediated tumor killing.
FLT3L, poly I:C, and anti-PD-L1 mAb overcome resistance to BRAFi
In addition to high numbers of activated DCs to promote efficient antigen presentation and T cell priming, a strong anti-tumor response requires increased recruitment of T cells that can be activated to become effector cells. When 4-HT-induced melanoma bearing mice were treated with BRAFi, a small molecule inhibitor of BRAFV600E, T cell infiltration increased only marginally. When BRAFi was combined with FLT3L + poly I:C treatment, however, T cell infiltration increased significantly. The T cells recruited following BRAFi + FLT3L + poly I:C treatment were cytolytic CD8+, IFN-g+, TNFa+ cells, and their ratio relative to regulatory T cells (Treg) was significantly increased. Therefore, a combination of BRAFi and FLT3L + poly I:C is highly effective in improving cytolytic effector T cells infiltration into BRAFV600E myeloma, and reducing tumor growth.
One side effect of the poly I:C treatment was increased PD-L1 expression in CD103+ DCs within the tumor. Based on improved CD103+ DC-mediated tumor responses to anti-PD-L1 treatment in initial experiments, B16 tumor-bearing mice were treated with a combination therapy (“tritherapy”) consisting of FLT3L + poly I:C + anti-PD-L1 mAb. Mice treated with this tritherapy showed significantly reduced tumor growth compared to treatment with FLT3L + poly I:C or anti-PD-L1 mAb alone. The tritherapy also increased recruitment and infiltration of cytolytic CD8+, IFN-g+, TNFa+ cells into the tumor and produce a higher CD8:Treg ratio over the other treatments. The enhanced antitumor efficacy with tritherapy in the B16 model encouraged the investigators to determine if this combination could help overcome resistance and relapse following BRAFi treatment alone. When 4-HT-induced melanoma mice were treated with BRAFi and tritherapy, tumor growth was reduced even more significantly compared with mice treated with FLT3L + poly I:C or anti-PD-L1 mAb alone. Importantly, a second experiment was performed in which BRAFi and tritherapy-treated mice were given a second application of 4-HT on the opposite flank. Thirty days later, these mice had significantly smaller tumors than tumor-bearing mice that were initially treated with just anti-PD-L1 mAb or FLT3L + poly I:C. This experiment demonstrated that the combinatorial therapy was superior in promoting T cells with a more lasting cytotoxic response memory.
The research reported in the Immunity study systematically teased apart the important immune cells that are involved in both the response, and lack of response, to melanoma. They also developed highly effective in vivo approaches to improve immune cell infiltration, antigen presentation, and cytotoxic activity that significantly diminished tumor growth. These results highlight the complexity that likely will be necessary to treat cancers effectively and also suggest that carefully designed combinatorial treatment approaches are likely to achieve the greatest benefits for cancer patients.