Taking a BiTE out of cancer
February 17, 2016
A bispecific T cell engager (BiTE) against Wilm’s tumor protein 1
Immuno-oncology - stimulating a patient’s own immune system to kill tumors - is emerging as a powerful strategy in the fight against cancer. Researchers at Memorial Sloan Kettering (Dao et al, 2015) engineered a hybrid antibody-like complex with dual specificities, which they called a bispecific T cell engager (BiTE). One end of the BiTE is specific for CD3, which is found on the cell membranes of CD4 and CD8 effector T cells. The opposite end of the molecule has a single chain fragment variable region (scFv) that binds to a specific peptide from the intracellular tumor-associated protein, Wilm’s tumor protein 1 (WT1). The scFv region came from a monoclonal antibody, ESK1, that recognizes the WT1 epitope RMF in the context of the antigen presenting molecule HLA-A*02:01. The ESK1-BiTE brings effector T cells that do not necessarily carry tumor antigen-specific T cell receptors into direct engagement with WT1-positvie tumors, stimulating the T cells to destroy the tumor cells.
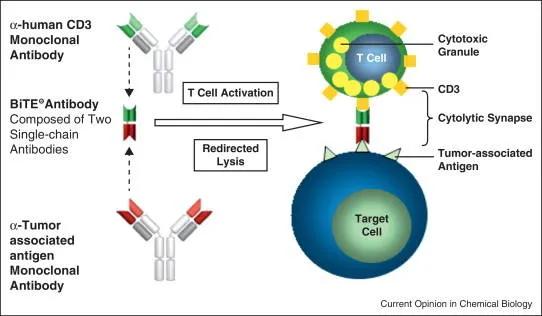
In vitro validation of ESK1-BiTE specificity and tumor cytotoxicity
Initial experiments with the ESK1-BiTE were performed in vitro by incubating human peripheral blood mononuclear cells (PBMCs) or isolated CD3-positive T cells with tumors known to express WT1 and HLA-A*02:01. These tumors included two human cell lines, SET-2 AML (acute myeloid leukemia) and BV173 Ph+ ALL (acute lymphoid leukemia), and a primary human ALL. The negative control was HL-60 AML, a cell line that is WT1-positive, but negative for HLA-A*02:01, which is required for the correct cell surface presentation of the peptide recognized by ESK1-BiTE. Liquid cultures were treated with either ESK1-BiTE or a control BiTE. PBMCs or CD3 T cells cultured with SET-2 AML cells and ESK1-BiTE demonstrated cytotoxic activity as measured by increased expression of the degranulation marker CD107, the lymphocyte activation marker CD137, and the pro-inflammatory molecules IFN-γ and TNF-α. Direct, targeted killing of the tumor cells was measured by either 51Chromium release or lactate dehydrogenase release assays. Targeted killing was only observed in ESK1-BiTE-treated cultures with tumor cells that expressed both WT1 and HLA-A*02:01. As expected, the ESK1-BiTE did not induce cytotoxicity when delivered to HLA-A*02:01-negative HL-60 AML cell cultures.
The Sloan Kettering investigators next performed similar experiments using T cells primed with B cells transduced with Epstein-Barr virus (EBV). Priming the T cells with EBV generated a population of T cells with specificity to viral antigens. These mature effector T cells could then be adoptively transferred to immunodeficient NSG™ mice, strain NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (005557), without the risk of the cells mounting a xenogeneic graft-versus-host (GvH) response. Prior to testing the efficacy of ESK1-BiTEin vivo, in vitro assays were needed to ensure that the ESK1-BiTE could redirect the EBV-primed T cells to attack the WT1-positive tumors. Cytotoxic lysis assays demonstrated that the EBV-primed T cells treated with ESK1-BiTE could target and kill a primary ovarian cancer, BV173 Ph+ ALL, and JMN mesothelioma. Collectively, these in vitro experiments demonstrated that the ESK1-BiTE could redirect antigen-primed T cells toward WT1-positive tumors in the context of HLA-A*02:01.
ESK1-BiTE demonstrates preclinical efficacy
The next step in evaluating the ESK1-BiTE was to show efficacy in vivo using immunodeficient NSG™ mice. Four cohorts of NSG™ mice were transplanted with SET-2 AML cells transduced with a luciferase reporter. The reporter allowed tumor cell tracking and measuring changes in tumor burden by bioluminescent imaging. Cohort one received only tumor cells. The other three cohorts received both tumor cells and EBV-primed PBMCs. Cohort two received no further treatment and served as a control for PBMC-mediated changes in tumor burden. Cohort three received a control-BiTE, and cohort four was treated with ESK1-BiTE. As expected, cohorts one and two showed identical tumor burden out to 18 days. The control-BiTE group showed a slight reduction in tumor burden at 15 days, but by 18 days, their tumor burden was the same as that observed in cohorts one and two. Only ESK1-BiTE-treated cohort 4 showed a significant reduction in tumor burden after 18 days. Further, all of the ESK1-BiTE-treated mice were alive at 30 days, by which time all of the mice in the other cohorts had succumbed. Moreover, the ESK1-BiTE-treated mice showed no signs of xenogeneic GvH disease even at 30 days-post engraftment. These experiments clearly demonstrated that the ESK1-BiTE could induce donor T cell-mediated killing of AML in vivo.
The Sloan Kettering team performed two additional in vivo experiments using primary human ALL and JMN mesothelioma to demonstrate the broader efficacy of ESK1-BiTE against other WT1-expressing human cancers. In both studies, the ESK1-BiTE significantly reduced tumor burden in the tumor-engrafted NSG™ hosts. These experiments provide unequivocal preclinical evidence that ESK1-BiTE can induce human T cell-mediated cytotoxicity to multiple WT1 expressing tumors in a mouse model of human disease.