NK cell mediated therapy for AML in NSG™ mice
December 9, 2015
Cellular therapy for AML
Researchers and clinicians are increasingly turning to immune cell based adoptive transfer therapies to treat both liquid and solid tumors. Young Acute Myeloid Leukemia (AML) patients treated with haploidentical hematopoietic stem cell (HSC) transplantation demonstrate prolonged survival and diminished rate of relapse. These patients show significant alloreactivity to the AML by donor-derived Natural Killer (NK) cells, making these cells strong candidates for adoptive therapy. A recent publication in the journalPLOS one (Cany et al., 2013) reports preclinical data showing the ability of umbilical cord blood (UCB)-derived HSC to generate NK cellsin vitro that, when transplanted into NSGTM mice bearing AML, diminished tumor burden and prolonged survival.
In vivo biodistribution and survival of human NK cells
Cany et al. evaluated human NK cell mediated adoptive therapy by examining thein vivo tissue distribution and survival of these cells following injection into highly immunodeficient NSGTM mice, strain NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (005557). In order to do so, they needed large numbers of cells and a method to track them. The authors isolated CD34+ HSC from a frozen sample of UCB and placed them in culture to expand the HSC. Using a method reported in a prior publication, the authors differentiated the UCB-HSC into UCB-NK cells and then expanded them in specialized medium along with IL-2, IL-7, IL-15, and SCF. The method generated CD3- CD56+ UCB-NK cells at >90% purity.
In order for the ex vivo expanded UCB-NK cells to home to the proper tissues in vivo, they must express the appropriate chemokine receptors and adhesion molecules. Evaluation of the cells by flow cytometry showed 52% of the cells expressed CCR6, 62% expressed CXCR3, and 10-20% expressed CXCR4. Chemokine mediated cell migration was validated by placing the UBC-NK cells in 5µm pore transwells with the chemokines CCL20, CXCL10, CXCL11, or CXCL12 (ligands specific for the receptors found on the UCB-NK cells) in the lower chamber of the culture well. Migration of UCB-NK cells to the lower chamber was confirmed by flow cytometry for CD56+ cells. Importantly, 10-20% of the UCB-NK cells also expressed the adhesion molecule CD62L (L-selectin). This data demonstrated theex vivo expanded cells expressed proteins necessary for homing and engraftment.
The in vivo biodistribution of the UCB-NK cells was performed by radio-labeling the cells with 111Indium. Following adoptive transfer into NSGTM mice, the cells were tracked using single photon emission tomography (SPECT) whole body imaging. In the first hour post intravenous delivery of the labeled cells, nearly all cells were found in the lungs of the mice. After 24 hours, the cells migrated to the spleen, bone marrow, and liver, with a few cells remaining in the lung. Tissues were harvested and flow cytometry was used to validate the presence of viable donor human UCB-NK cells (and not just radio-labeled cell fragments).
The remaining question was survivability of the cells. In prior clinical reports demonstrating NK cell tumor responses, the patients showed short-term elevation of endogenous IL-15. Therefore, the authors examined survival and expansion of UCB-NK cells following adoptive transfer in mice treated with PBS (phosphate buffered saline) or human IL-15. Mice treated with PBS lost UCB-NK cells in blood, spleen and bone marrow by 7 days post injection. In contrast, mice treated with IL-15 retained donor cells in these tissues at day 7, the cells expanded at day 14, and they increased expression of the NK cell maturation marker CD16.
These data clearly showed the ability to generate large numbers of human NK cellsex vivo and these cells were capable of homing to the appropriate tissues in vivo in an immunodeficient mouse. Inclusion of exogenous human IL-15 improved survival, expansion and maturation of the human cells. Also, SPECT imaging can be useful to track homing of NK cells during both pre-clinical and clinical testing.
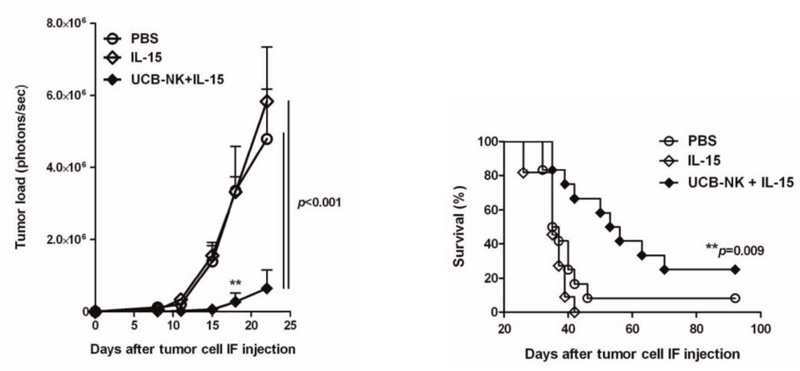
Fig 1. UCB-NK cells diminish tumor burden and increase survival in NSGTM mice engrafted with human AML. (Left) NSG™ mice treated with a combination of IL-15 and UCB-NK cells show significantly delayed and decreased tumor burden compared to controls treated with either PBS or IL-15 alone. Tumor load was determined by bioluminescent imaging of luciferase labeled K562 AML cells. (Right) Treatment with IL-15 and UCB-NK cells prolonged survival of NSG™ host mice compared to PBS and IL-15 controls.
(Left) NSG™ mice treated with a combination of IL-15 and UCB-NK cells show significantly delayed and decreased tumor burden compared to controls treated with either PBS or IL-15 alone. Tumor load was determined by bioluminescent imaging of luciferase labeled K562 AML cells. (Right) Treatment with IL-15 and UCB-NK cells prolonged survival of NSG™ host mice compared to PBS and IL-15 controls.
In vivo targeting of AML by NK cells
The next set of experiments were designed to determine if the UCB-NK cells could recognize AML targetsin vivo, diminish tumor burden, and increase survival of the recipients. For the AML target, Cany et al. used the K562 cell line, which is routinely used in NK cell killing assays. The K562 cells were transduced with a lentiviral vector carrying luciferase and GFP driven by a CMV promoter. Labeled AML cells (K562.LucGFP) were then tracked and quantitated using bioluminescent imaging (BLI). NSGTM mice were injected with 105 K562.LucGFP directly into the right femur. One day later, one cohort of mice were treated with PBS alone, a second cohort were treated with human IL-15 alone every 2-3 days for 14 days, and a third cohort were treated with a single injection of 20x106 UCB-NK cells plus the same regimen of human IL-15 as in the second cohort. BLI was performed beginning at day 8, then every 3-4 days for two weeks. Survival of the mice was followed out to 90 days. Mice treated with either PBS or IL-15 alone showed presence of AML in femur by BLI beginning at day 15. Tumor load in these two control groups expanded at similar rates out to day 25. Nearly all of the mice in these two groups succumbed to disease by 40 days. In contrast, 8 out of 12 mice in the IL-15 plus UCB-NK cell tested group had no detectable tumor in femurs at day 15. BLI did detect signal at subsequent time points but signal was significantly less than signal found in the control groups. Prolonged survival was also observed. Twenty-five percent of the mice treated with IL-15 and UCB-NK cells survived out to 90 days.
In conclusion, Cany et al. were able to generate an expanded pool of human NK cells derived from UCB. These cells retained homing and adhesion molecules that allowed them to engraft appropriate tissuesin vivo. The UCB-NK cells were able to recognize and reduce AML tumor cells in a preclinical mouse model of human disease, leading to prolonged survival of host animals. The data provided in this report provide important preclinical evidence supporting the use of adoptive immunotherapy to treat human cancer.