IL-12-induced anti-tumor immunity in Humanized Mice
eNews | November 11, 2015Targeted cytokine therapy for cancer
Research exploring immune system modulators to treat cancer is leading to exciting and highly encouraging new clinical therapeutic approaches. Mice reconstituted with a human immune system and engrafted with human tumors are emerging as a powerful new preclinical tool to evaluate new therapeutic strategies. A new publication in the journalOncoImmunology (Schilback et al., 2015) reveals that tumor-targeted IL-12 coupled with IL-2 or IL-7 stimulates IFN-g and TNF-dependent tumor dormancy. Tumors treated with IL-12 and IL-2 show enhanced cytotoxic T and NK cell responses, tumor regression, and 100% host survival. Humanized tumor-bearing mice allow preclinical testing of human immune-modulatory therapeutics, and may accelerate the latters’ translations to clinical trials.
Humanized tumor-bearing mice for immuno-oncology
Much to the surprise of immunologists, humanized mice engrafted with non-HLA matched tumors do not appear to mount an allogeneic tumor rejection response. The survival of HLA-mismatched tumors likely results from the myriad of mechanisms that tumors utilize to evade host immunity. This new platform allows researchers to test,in vivo, engineered biomolecules that stimulate the immune system to engage and kill tumor cells and to dissect the associated mechanisms of action. Schilback and colleagues used highly immunodeficient NSG™ mice, NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (005557), humanized by engrafting bone marrow-derived CD34+ hematopoietic stem cells (hHSCs) obtained from G-CSF-mobilized blood. Following immunologic reconstitution of the host (by ~12 weeks), the mice were engrafted subcutaneously with a human allogeneic rhabdomyosarcoma (RMS) cell line (A204). Tumors were established by three weeks, and untreated mice typically survived for only ~50 days following tumor engraftment.
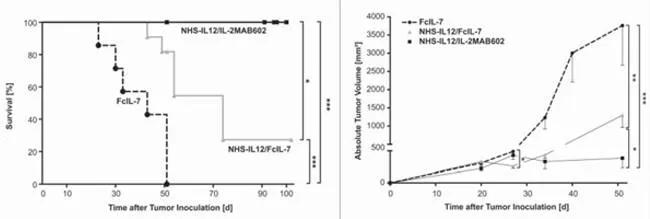
Tumor-targeted IL-12 in combination with IL-2 increased survival and decreased tumor volume in humanized NSG™ mice engrafted with human rhabdomyosarcoma. (Left) Mice treated with a combination of IL-12 (NHS-IL12) and IL-2 (IL-2MAB602) maintained 100% survival out to 100 days. NHS-IL12 with FcIL-7 improved survival and FcIL-7 alone had no effect. (Right) Treatment with NHS-IL12 and IL-2MAB602 prevented tumor growth, NHS-IL12 and FcIL-7 significantly suppressed tumor growth, and FcIL-7 alone had no effect.
(Left) Mice treated with a combination of IL-12 (NHS-IL12) and IL-2 (IL-2MAB602) maintained 100% survival out to 100 days. NHS-IL12 with FcIL-7 improved survival and FcIL-7 alone had no effect. (Right) Treatment with NHS-IL12 and IL-2MAB602 prevented tumor growth, NHS-IL12 and FcIL-7 significantly suppressed tumor growth, and FcIL-7 alone had no effect.
Tumor-targeted delivery of IL-12
IL-12 normally is produced by activated antigen presenting cells (dendritic cells and macrophages) and is associated with promoting Th1 effector T cell function and NK cell activation. IL-12 also plays a role in polarizing macrophages to the pro-inflammatory M1 state versus the anti-inflammatory M2 state that promotes tissue repair. Prior studies with IL-12- treated tumors provided evidence for immune activation and tumor shrinkage, but systemic delivery had toxic consequences. Therefore, Schilbach and colleagues developed a strategy for delivering IL-12 directly to the tumor by creating a fusion complex between an antibody (NHS76) that binds naked histone DNA complexes and IL-12 (NHS-IL12). Because necrotic areas within tumors have exposed naked histone DNA complexes, this NHS-IL12 complex could specifically target the tumor. Further, IL-12 in the NHS-IL12 complex had a longer efficacious half-life compared with native IL-12 and decrease systemic toxicity. Targetedin vivo delivery of NHS-IL12 to the RMS tumors was confirmed using 123I-labeled NHS-IL12 imaging.
Differential tumor killing mechanisms with IL-12 and IL-2 versus IL-12 and IL-7
In order to maximize anti-tumor T cell responses in the humanized and RMS-engrafted mice, Schilbach and colleagues combined NHS-IL12 treatment with either IL-2 or IL-7. IL-2 was complexed with the monoclonal antibody MAB602, creating 2MAB602. This antibody/IL-2 complex promotes greater bioavailability and improved IL-2 effector functions. IL-7, a T cell survival factor, was administered as recombinant IL-7 (FcIL-7). In a control experiment, FcIL-7 treatment alone, without NHS-IL12, failed to suppress tumor growth in humanized/RMS tumor engrafted mice; indeed, tumors in this group grew 6.5-fold between days 27 and 52. Further, 4 of 7 mice did not survive to the 52 day end point. In contrast, 2 of 11 mice treated with NHS-IL12 and FcIL-7 survived to 52 days, and tumor growth was significantly suppressed. Finally, NHS-IL12 combined with 2MAB602 completely prevented tumor growth and 100% of the treated cohort survived. In fact, when mice were treated with NHS-IL12/2MAB602 for longer, tumors were eliminated in 3 out of 4 treated mice, and tumor growth was suppressed in the 4th.
The authors next examined the tumors post-treatment for immune cell infiltrates in order to investigate the immune cell responses that were responsible for the observed tumor regression. The control FcIL-7-treated tumors were infiltrated with CD56+ NK cells and CD68+ macrophages and were largely devoid of CD4+ and CD8+ T cells. In contrast, the NHS-IL12/FcIL-7 and NHS-IL12/2MAB602 treated tumors were highly infiltrated with all four of these human immune cell populations. The tumors treated with NHS-IL12/FcIL-7 and NHS-IL12/2MAB602 also showed significantly increased staining for HLA-DR, a class II antigen presentation marker found on macrophages, suggesting pro-inflammatory M1 macrophages were enhanced. Examination of tumor -derived NK cells from all three treatment groups showed expression of the maturation markers NKG2D and DNAM-1. NHS-IL12/FcIL-7 treated NK cells also expressed NKG2E, NKp44, and NKp46, and the NHS-IL12/2MAB602 treated NK cells expressed NKp30. Together, these data demonstrated differential functional maturity of the NK cells that were associated with each cytokine treatment. Another major difference in the various treatment groups was expression of perforin and granzyme K, which was found only in the tumors treated with NHS-IL12/2MAB602. Perforin and granzyme K are produced by cytotoxic T cells and NK cells. These differential cell surface markers and secreted proteins in each treatment group suggests that the mode of tumor growth arrest and killing by the immune cells was established by disparate mechanisms. Examination of the proliferation markers PCNA and Ki67 and the cell cycle senescence markers p-HP1g and p16INK4a in the tumors demonstrated low proliferation and increased senescence signatures in both IL-12 treatment groups. In addition, NHS-IL12/FcIL-7 -treated tumors expressed high levels of CD161 and low expression of RORC, indicating that the infiltrating T and NK cells do not have lytic activity, but secrete high amounts of IFN-g and TNF, two factors known to induce senescence. CD161 or RORC was not detected at all in tumors treated with NHS-IL12/2MAB602, confirming the presence of T and NK cells with more lytic characteristics. Therefore, while IL-12 promoted the maturation of human immune tumor infiltrating cells that promoted senescence, combining the treatment with IL-2 promoted the development of cells with enhanced cytotoxic activity.
Overall, the authors concluded that without IL-12 treatment, the human cells infiltrating the RMS tumors were immature NK cells and HLA class II-negative M2 macrophages that were permissive for tumor proliferation. Treatment with IL-12 drove development of M1 pro-inflammatory macrophages and up-regulated several receptors on NK cells required for their functionally maturity and ability to secrete IFN-g and TNF. The combination of IL-12 and IL-7 suppressed tumor cell proliferation but did not promote the human cytotoxic immune cell responses. Combining IL-12 with IL-2 also led to tumor senescence, but the infiltrating T and NK cells demonstrated cytotoxic activity that killed tumor cells and allowed 100% of the treated mice to survive. The tumor-bearing humanized mouse provides an excellent platform for evaluating the efficacy of biomolecules engineered to enhance anti-tumor human immune responses.