Intestinal cell modifications aid gut microbiota during infection, help host
October 26, 2014
The gut microbiota
Each of us has a unique collection of microorganisms in our intestine, known as the gut microbiota, whose physiological functions include helping food digestion, producing vitamins B and K, and maintaining the intestinal mucosa. A recent publication in Nature (Pickard et al., 2014) reveals that the gut microbiota can also be selected by the host to enhance its tolerance to systemic infection.
Fucosylation feeds the microbiota during systemic infection
It is known that systemic infection induces conserved responses that render both resistance and tolerance to infection (Ayres et al., 2012). Further, these responses can be modulated by reduced food consumption (anorexia) (Ayres et al., 2009; Murray et al., 1979). Because the host-derived sugar fucose is accessible to the microbiota but not to the host for energy consumption (Bocci et al., 1969; Becker et al., 2003), Pickard et al., hypothesized that the host might utilize fucose to support beneficial microbiota during the acute phase of pathogenic bacterial infection. Symptoms of systemic bacterial infection, including anorexia, were induced by administering bacterial-derived molecules, including Toll-like receptor agonists such as lipopolysaccharide (LPS). The investigators found that, under normal conditions, the small intestine of specific pathogen-free (SPF) mice is largely free of surface fucose, but following the systemic injection of LPS, rapid and sustained glycoprotein fucosylation is observed throughout the organ. These fucosylated glycoproteins are then released into the intestinal lumen where they are consumed by the resident microbiota. Similar results were observed in multiple inbred strains, including BALB/cJ (000651), C3H/HeJ (000659), C57BL/6J (000664) and NOD/ShiLtJ (001976), suggesting that genetic background influences these changes only minimally, if at all.
Fucosylation inhibits bacterial invasion of intestinal tissues
Pickard et al. undertook additional studies to elucidate the mechanism of LPS-stimulated fucosylation and its role in host defense. Specifically, they looked at the LPS-dependent regulation ofFut2, which encodes a specific fucosyltransferase that mediates cell surface molecule fucosylation. Their data shows thatFut2 is expressed in intestinal epithelial cells (IECs) following systemic administration of LPS and suggests that LPS-stimulated Fut2 up-regulation occurs via a lymphocyte-mediated pathway (Fig. 1)
1) LPS stimulates dendritic cells to release interleukin-23 (IL-23).
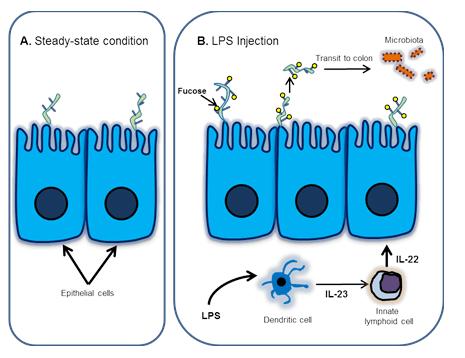
A. In steady-state conditions, fucosylation is not induced in the small intestine.
B. Signaling pathway leading to fucosylation and maintenance of the gut microbiota following LPS injection.
2) IL-23 activates innate lymphoid cells to secrete IL-22.
3) IL-22 in turn, induces Fut2 expression in IECs (Fig.1).
Pickard et al. demonstrated that gene deletion of Fut2 prevents fucosylation in IECs in response to LPS, indicating that Fut2 expression is required for the LPS-stimulated fucosylation seen in IECs following systemic infection. Moreover, they demonstrated that mice lacking Fut2 recover from weight loss more slowly following LPS-induced anorexia than their littermate controls’, suggesting that fucosylation is beneficial to the host under such stressful conditions. This beneficial effect is dependent on the microbiota, because microbiota-depleted (germ-free) mice show impaired weight gain following LPS-induced anorexia, despite normal IECs fucosylation.
Finally, to determine whether fucosylation improves the host’s fitness during infection, Pickard et al. infected Fut2-deficient and control mice with the non-lethal intestinal pathogen,Citrobacter rodentium, prior to LPS administration. They found that the C.rodentium-infected, Fut2-deficient mice lose significantly more weight and have more pronounced colonic hyperplasia (a trademark of this infection) compared withFut2-sufficient control mice, indicating that inducible fucosylation by Fut2 is crucial for limiting colonic cell proliferation and contributes to improved host fitness during infection.
The most exciting aspect of this work might be what it contributes to our understanding of beneficial interactions between the microbiota and its host. There is growing awareness of host factors, cells and environmental conditions that shape the microbiota. Little is known, however, about whether the host can select for a beneficial microbiota to increase its own fitness under adverse conditions (Rakoff-Nahoum and Comstock, 2014). The studies by Pickard et al. reveal a mechanism by which such selection may occur.