Transducing human stem cells to drive tumor-specific TCR expression
An important advance in developing safer gene therapies for immune-mediated cancer treatments has been reported in Cancer Research (Gschweng et al. 2014). The safety of viral vector delivery of genes into human hematopoietic stem cells (HSC) is a legitimate concern based on adverse events observed in early clinical trials. Gschweng et al. have addressed this issue by creating a vector engineered to co-deliver a suicide gene along with a therapeutic anti-tumor T cell receptor (TCR). The suicide gene allows elimination of engineered T cells that promote autoimmune or oncogenic responses. Functional evaluation of the transduced human HSCs was achieved using immune-deficient NSG mice expressing an HLA class I transgene. The suicide gene has an added advantage of also being capable of modifying substrates that enable tracking of gene-modified cells in vivo by positron emission tomography (PET).
Genetically engineered HSC for cancer treatment
There are several approaches to develop therapies using cytotoxic T cells (CTLs) specific for tumor antigens. Although in vitro expanded, mature, tumor-specific CTLs show efficacy in tumor killing following engraftment, they do not have long-term self-renewal capacity and do not lead to life-long immunity. Viral vector-mediated gene delivery to HSCs solves this problem, but holds the following inherent risks of its own:
- Vector-associated promoters may activate proto-oncogenes near the insertion site
- Engineered TCRs may induce autoimmunity if directed to self-antigens shared by the tumor and other, normal host cells.
Therefore, a method to eliminate transduced cell populations if an adverse response develops in a treated patient would be an important advance allowing for safer clinical trials.
Gschweng et al. included the suicide gene, sr39TK, in their lentiviral construct to allow both the elimination of transduced cells and imaging by PET. The sr39TK gene encodes a modified herpes simplex virus thymidine kinase (HSV-TK). The drug ganciclovir is an antiviral compound that is phosphorylated by HSV-TK and selectively inhibits DNA replication. In addition, sr39TK modifies radiolabeled substrate [18F]-FGBH to allow non-invasive, in vivo imaging to track engraftment, expansion, and homing of the successfully targeted HSCs. The gene therapy construct also expresses a TCR in developmentally mature CD8+ CTLs specific for the well-characterized NY-ESO-1 peptide, an antigen that is expressed both in normal testis and various human tumors. The peptide is presented by HLA class I, allowing recognition and killing by CTLs that express the appropriate TCR. Importantly, the sr39TK gene was engineered into the vector downstream of the TCR alleles such that HSV-TK expression is driven specifically in transduced, TCR-expressing CD8+ CTLs.
Gschweng et al. tested their gene therapy vector by transducing human CD34+ HSCs obtained from G-CSF-mobilized peripheral blood cells (PBSCs). The PBSCs were transduced with the therapeutic lentivirus and were transplanted into myeloablated neonatal mice by intrahepatic injection (see figure). The mouse strain used as the host for these cells was NOD.Cg-Prkdcscid Il2rgtm1Wjl Tg(HLA-A2.1)1Enge/SzJ (009617), commonly called NSG-A2.1. This strain is based on the highly immunodeficient NOD scid gamma (NSG, 005557) mouse that were bred to carry a transgene expressing the human leukocyte antigen (HLA) class I subtype A2.1.
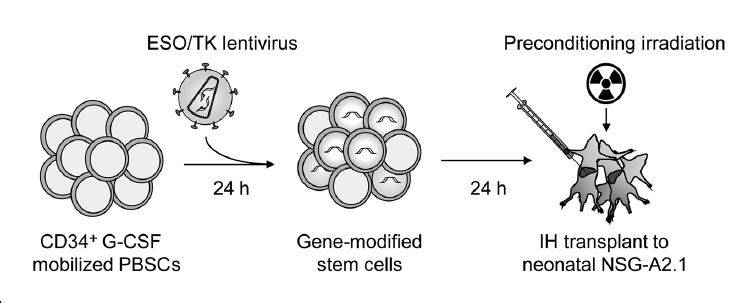
Experimental design for testing gene-modified human stem cells. Human CD34+ hematopoietic stem cells were obtained from G-CSF-mobilized peripheral blood (PBSC), transduced with a lentivirus encoding sr39TK and the NY-ESO-1 specific TCR, and then transplanted into myeloablated neonatal NSG-A2.1 (009617) mice by intrahepatic injection.
Human CD8 T cells in NSG-A2.1 hosts express TCR specific for tumor antigen
The investigators used flow cytometry to evaluate recipients for donor HSC engraftment and expression of the engineered TCR in human CD8+ T cells. All recipients engrafted with donor human HSCs, but only 8 of 15 mice expressed the NY-ESO-1-specific TCR in CD8+ T cells. The NY-ESO-1 TCR was not expressed in CD4+ T cells. CD8+ T cells expressing NY-ESO-1 were obtained from two recipients, expanded in culture, and effector function was measured by a radioactive chromium release cytotoxicity assay using human melanoma cells expressing the antigen as target cells. Specific target cell lysis (measured by chromium release) and CTL-derived IFNγ secretion was observed only in cultures containing target cells expressing both HLA-A2.1 and the NY-ESO-1 antigen.
In vivo detection and ablation of donor transduced T cells
Mice engrafted with transduced human PBSCs were injected with [18F]-FGBH and imaged with PET. As expected, background signal is observed in the abdominal area in non-humanized and humanized, but non-transduced, mice due to probe elimination throughout the gastrointestinal tract. In mice humanized with transduced HSCs, signal is observed in the long bones, sternum, thymus and vertebrae − all sites expected to be repopulated with donor hematopoietic cells.
Next, two cohorts of humanized mice were prepared, one with non-transduced PBSC and the other with transduced PBSC. Both groups were injected with ganciclovir for 5 days and imaged by PET one week later. Only the mice transduced with sr39TK were completely devoid of PET signal. Flow cytometry of the two ganciclovir-treated groups revealed no significant differences in overall donor cell engraftment and similar percentages of CD3+ T cells and CD19+ B cells. Only the transduced PBSC-engrafted group showed loss of CD8+ T cells.
Gene therapy with a safety net
In conclusion, Gschweng et al. clearly demonstrate that human HSCs can be transduced with a lentiviral vector delivering both a therapeutic antigen-specific TCR and a suicide gene capable of providing both in vivo validation of engraftment and donor cell tracking. Donor-derived T cells show tumor antigen-specific killing of target cells and those same cells can be eliminated from circulation by ganciclovir treatment. This elegant vector design should be useful in a wide range of applications, and will provide a unique safety net during clinical trials to reduce adverse, vector-dependent responses in patients already in dire health.