Hair cell loss causes deafness in mice
JAX Notes | October 28, 2009Although hereditary deafness is a common human birth defect, the majority of genes that underlie hearing disorders in humans are thought to be unidentified. One of those genes has just been discovered. A research team headed by Dr. Yoko Nakano at the University of Iowa recently reported that a defect in the claudin9 gene (Cldn9), which encodes a previously uncharacterized inner ear protein, destroys cochlear hair cells and causes deafness in mice (Nakanoet al. 2009). Because the amino acid sequence of claudin9 is highly conserved in mice and humans, similar defects may also cause hearing loss in humans.
How you hear
To function properly, your ears depend on sheets of epithelial cells to control the diffusion of K+ and Na+ ions between two compositionally distinct inner ear fluids, the endolymph and the perilymph. The endolymph bathes the stereocilia of the cochlear sensory hair cells and has a high K+ concentration and a low Na+ concentration (approximately 140 mM and 1 mM respectively). In contrast, the perilymph bathes the basolateral side of hair cells and has a low K+ and a high Na+ concentration (approximately 2 mM and 140 mM respectively). The differential K+ and Na+ concentrations generate an electrical potential or polarity of approximately 80 mV between the endolymph and perilymph. Maintaining that polarity is the job of epithelial cell membrane structures called tight-junctions. Tight-junctions prevent diffusion of K+ from the endolymph to the perilymph and the basolateral sides of the hair cells. When sound waves reach the inner ear, they deflect the stereocilia, and K+ and other cations enter the hair cells. As a result of the ensuing depolarization, neurological signals are transmitted to the brain, and you "hear" a sound. The resting polarity is restored when the sound dissipates.
The permeability of tight-junctions varies greatly from tissue to tissue and depends on the expression patterns of tight-junction proteins called claudins, 24 of which have been identified in humans.
A mouse mutation reveals a protein's unsuspected function
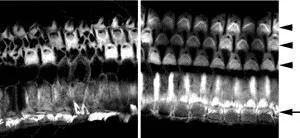
The mutant mouse used by Nakano and her team, C57BL/6J-Cldn9nmf329/J, was produced in an ethylnitrosourea (ENU) mutagenesis project at The Jackson Laboratory. Before Nakano and her team published their research, the mouse was named C57BL/6J-nmf329/J, "nmf329" indicating that the mouse was the 329th mutant produced at the JAX neuromutagenesis facility. JAX scientists had roughly mapped the deafness-causing mutation to Chr 17. Nakano and her team narrowed that location to a smaller region containing 73 genes, none of which had ever been associated with deafness. One of the genes was Cldn9. Claudin9 is the most highly expressed claudin in the inner ear, occurring in all of the major epithelial cell types that enclose the endolymph. Its biological function was unknown.
Nakano and her team found that the Cldn9 gene in the C57BL/6J-Cldn9nmf329/J mouse has a T-to-C point mutation 102 bases downstream of the start codon. Their suspicion that this mutation was responsible for the mouse's deafness was confirmed by several lines of evidence: 1) the K+ concentration in the perilymph of the mouse is abnormally high, suggesting defective tight junctions; 2) whereas wild-type cultured cells maintain the Na+ and K+ barrier, mutant cells do not; 3) hair cells inCldn9-defective cochlea are rescued in vitro when cultured in a low-K+ medium; and 4) the hair cells are not destroyed in vivo in mice deficient for both Cldn9 and the POU domain, class 3, transcription factor 4 (Pou3f4) gene. (These double mutants, which were produced by crossing C57BL/6J-Cldn9nmf329/J and C3HeB/FeJ-Pou3f4del-J/J (004406) mice, have low endolymphatic K+ concentrations.) Essentially, the researchers found that whenCldn9 is mutated, potassium floods the wrong part of the sensory hair cells, killing most and leaving the remaining ones functionally defective.
The findings by Dr. Nakano and her team contribute significantly to hearing research. They reveal the biological significance ofCldn9 in hearing, give us new insights into how tight-junctions function in the ear, and help characterize a potentially very useful mouse model for hearing research.
References
Nakano Y, Kim SH, Kim HM, Sanneman JD, Zhang Y, Smith RJ, Marcus DC, Wangemann P, Nessler RA, Banfi B. 2009. A claudin-9-based ion permeability barrier is essential for hearing.PLoS Genet 5:e1000610. Epub 2009 Aug 21.