
For more than 90 years, research at JAX has expanded knowledge of mammalian genetics and contributed to medical progress. Now, at its Bar Harbor, Maine, and Farmington, Connecticut, research campuses, JAX is poised to bridge the gaps between biological knowledge and medical progress. With unmatched model organism research expertise and resources and modern research methods and tools in hand, JAX scientists are accelerating both basic discovery and clinical translation for patient benefit.
Researchers at JAX contribute to a highly impactful, multi-disciplinary biomedical research program focusing on mammalian genetics and human genomics. Faculty, trainees and laboratory staff benefit from collaborative intra-institutional science, a broad scope for faculty inquiry, robust scientific resources and advanced technologies, and extensive support services. The unique environment catalyzes discovery and insights across the spectrum of basic, translational and preclinical research.
Comprehensive sponsored research support yields high grant application success rates and thorough grant management.
Advanced equipment and deep expertise expand research possibilities.
Professional support is provided to faculty for day-to-day tasks as well as specialized ventures.



Mark Adams
Ph.D.
Professor & Interim Scientific Director, The Jackson Laboratory for Genomic Medicine
Meet Mark
Nadia Rosenthal
Ph.D., FMedSci, FAAHMS
Scientific Director and Professor, The Jackson Laboratory for Mammalian Genetics, the Maxine Groffsky Endowed Chair
Meet Nadia

I was attracted by how diverse the research programs are at JAX, beyond just cancer. I think it’s great to benefit from researchers with diverse expertise and backgrounds.
– Assistant Professor Eric Wang, Ph.D., leukemia researcher

Endometriosis research at JAX is improving the lives of those impacted by the disease – from seeking to develop early detection with genetic testing to understanding the development of the condition at a single cell level.
View more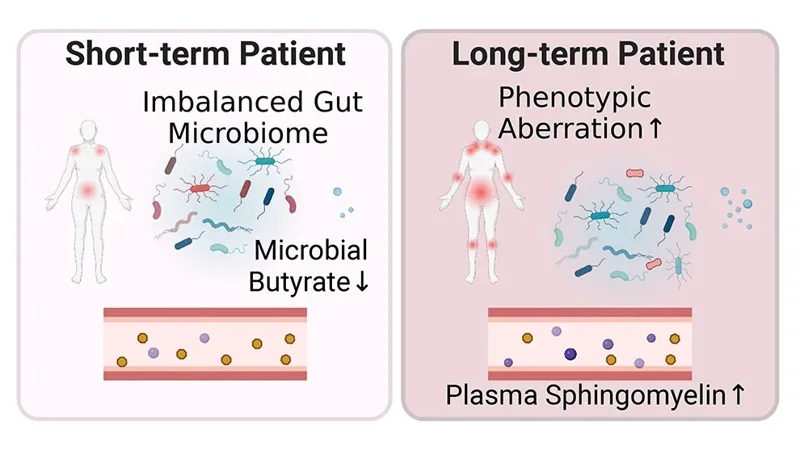
A new study from Oh, Unutmaz and their collaborators delves into host-microbiome interactions in ME/CFS as well as potential metabolic consequences.
View more
Combining genetic therapy and bone marrow transplantation may offer comprehensive treatment solution for multiple sulfatase deficiency.
View more