Depletion of Tregs Improves Anti-Tumor Immunity in Humanized Mice
July 6, 2016Tumors use multiple mechanisms to evade responses by the patient’s immune system. One mechanism involves regulatory T cells (Tregs). In a healthy individual, Tregs are an important immune cell population that helps to maintain immune tolerance and prevent autoimmunity. Treg infiltration of patient tumors, however, correlates strongly with poor survival because the Tregs suppress T effector cell (Teff) anti-tumor cytotoxic responses. Tumors attract Tregs by expressing the chemokine CCL22, which binds to CCR4 receptors on the Treg cell surface. In the May 2016 issue ofOncoimmunology investigators at Harvard Medical School in Boston and the University of Pennsylvania in Philadelphia show that monoclonal antibodies (mAb’s) to CCR4 can prevent Tregs from infiltrating tumors in vivo, thus enabling Teff-mediated anti-tumor immunity and suppression of tumor growth (Chang et al 2016).
Human immunosuppressive Tregs express chemokine receptor CCR4
The Harvard and UPenn investigative teams began their study by characterizing human peripheral blood mononuclear cells (hPBMC) for subsets of T cells that express CCR4. Tregs were defined by the markers CD4+CD25hiCD127dim/- and Teff defined by CD4+CD25-CD127+. Flow cytometry revealed that approximately 90% of these Tregs and approximately 10% (4-40%, expression was highly variable) of Teffs expressed CCR4 (CCR4+). When Tregs were defined as CD4+CD25hiFoxP3+, approximately 85% were CCR4+. QuantiBrite fluorescent beads were used to demonstrate that the density of CCR4 receptors on the Treg cell surface was ~2.5-fold greater than on the surface of Teffs. The investigators next examined whether CCR4+ versus CCR4- Tregs could suppress proliferation of phytohemagglutin (PHA)-stimulated, CFSE-labeled Teffs in vitro. Only co-cultures containing CCR4+ Tregs suppressed Teff proliferation. These experiments showed that hPBMC-derived Treg populations have both a higher frequency and a higher density of CCR4 receptors than other T cell populations, and that CCR4+ Tregs have immunosuppressive capacity.
Monoclonal antibodies to CCR4 block chemotaxis to CCL22 expressing ovarian tumors
The authors next sought to determine if mAb’s specific for CCR4 could interfere with chemotaxic migration toward a CCL22 source. Three human ovarian cancer (OvCA ) cell lines were identified – IGROV-1, OVCAR-5, and OVCAR-8 - that had high, medium, and no CCL22 expression, respectively. None of these OvCA cell lines expressed CCR4. Two anti-CCR4-specific mAb’s were tested: mAb2-3 IgG1 that was shown to havein vivo cell depletion activity and mAb2-3 IgG4 that does not induce depletion. Both mAb’s blocked in vitro migration of Tregs to IGROV-1 and OVCAR-5 cells, but not to OVCAR-8 cells. Both mAbs also inhibited Treg chemotaxis to recombinant human CCL22 in a dose-dependent manner.
To test the efficiency with which these mAbs block Treg migration in vivo, NSGTM mice (NOD.Cg- PrkdcscidIl2rgtm1Wjl/SzJ, 005557) were engrafted with high CCL22-expressing IGROV-1 cells followed by IV injection of luciferase-transduced, human CD4+CD25hiCD127dim/- Tregs. As shown in figure A, the luciferase-tagged Tregs migrated to and infiltrated tumors in the PBS-treated control mice. As expected, mice treated with mAb2-3 IgG1 lacked both circulating and tumor-associated Tregs due to depletion of CCR4+ cells. Mice treated with mAb2-3 IgG4, however, retained Tregs in circulation and did not accumulate labeled cells in the tumor. Importantly, mice injected with luciferase-transduced human CD4+ T cells and mAb2-3 IgG1 retained cells in circulation and in the tumor. Taken together, these experiments demonstrated that human Tregs migrate to tumors via CCL22/CCR4-mediated chemotaxis and that their migration can be inhibited both in vitro and in vivo using mAb’s specific for CCR4. CD4+ Teff cells persist in vivo following treatment with either mAb, making them available for antitumor immunity in the absence of Treg-mediated suppression.
A.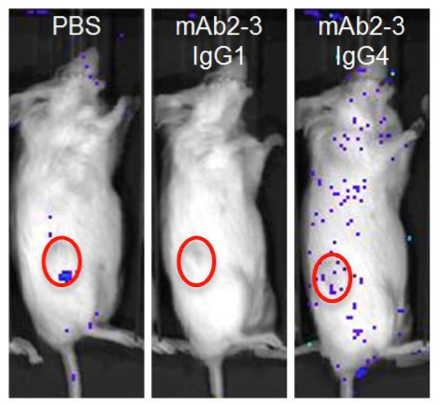 B.
B.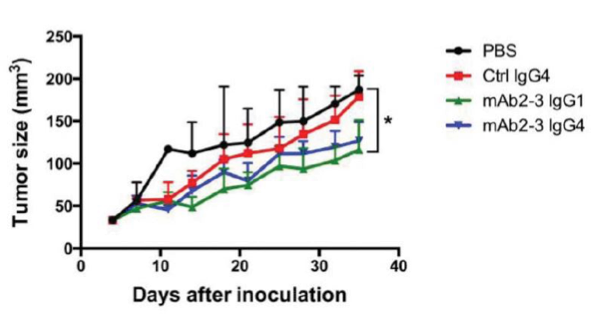
Depleting Regulatory T cells or blocking tumor infiltration with anti-CCR4-specific antibodies enhances T effector killing of ovarian cancer.(A) NSGTM mice subcutaneously engrafted in the flank with ovarian cancer cell line IGROV-1 (red circles). Mice were subsequently injected with luciferase tagged human CD4+ CD25+ CD127dim/- Tregs (~85% CCR4+). PBS control treated mice show homing of Tregs to the CCL22 chemokine-expressing tumor. Mice treated with mAb2-3 IgG1 are devoid of Tregs, and mice treated with mAb2-3 IgG4 lack homing of Tregs to the tumor. (B) Tumor size over time in IGROV-1 tumor engrafted NSGTM mice injected with IGROV-1 tumor primed human T cells and treated with either PBS, control IgG4, mAb2-3 IgG1, or mAb2-3 IgG4. Depleting Tregs with mAb2-3 IgG1 or blocking Treg homing to the tumor with mAb2-3 IgG4 allowed T eff-mediated suppression of tumor growth. Adapted from Chang et al, 2016.
Blocking tumor-infiltrating Tregs allows Teff-mediated reductions in tumor growth
Because mAb2-3 can block Tregs migrating toward OvCA, the next step was to determine if depleting Tregs would allow Teffs to suppress tumor growth. The Harvard and UPenn teams created tumor-primed T cells (TP-T) that specifically recognized IGROV-1 tumors by incubating human T cells with IL-2, IL-7, and autologous human dendritic cells (DCs) pulsed with a IGROV-1 tumor cell lysate. The TP-T cells produced IFN-g when cultured with tumor antigen-loaded DCs, but not when incubated with unpulsed DCs, demonstrating IGROV-1 specific Teff activity. Because the TP-T cell population contained approximately 32% CD4+ CD25+ CCR4+ Tregs, a second experiment was performed with a TP-T cell fraction that had been depleted of Tregs cells using mAb2-3-conjugated magnetic beads. The two samples were incubated with IGROV-1 cells to compare Teff activity. The Treg-depleted TP-T cells showed significantly greater IFN-g production when compared to the whole TP-T cell population, demonstrating improved IGROV-1 specific Teff activity in the absence of CCR4+ Tregs.
In vivo anti-tumor efficacy was tested by engrafting NSGTM mice with luciferase-tagged IGROV-1 cells and TP-T cells. Cohorts of mice were treated with either PBS, control IgG4, mAb2-3 IgG1, or mAb2-3 IgG4 twice weekly for 5 weeks. Tumor bioluminescence decreased the most over time in the mice treated with the Treg-depleting mAb2-3 IgG1, followed by treatment with mAb2-3 IgG4. The various treatments showed similar effects on tumor volume and tumor weight. The experiment was repeated using 2.5 fold more TP-T, and a similar response was observed. Tumors from these treatment groups then were examined using immunohistochemistry for CD3+ T cell and CD25+ T cell (Treg) infiltration. All treated tumors had similar levels of CD3+ T cell infiltration. The mAb2-3 IgG1- and mAb2-3 IgG4-treated tumors both showed reductions in CD25+ Treg infiltration. Examination of peripheral blood showed all treatment groups retained similar frequencies of circulating human CD4+ and CD8+ T cells. As expected, only the mAb2-3 IgG1 treated-mice showed reductions in circulating CD25+ Tregs.
Collectively, the in vivo experiments supported the hypothesis that tumor-infiltrating Tregs suppress anti-tumor immunity. Moreover, when the Tregs are either depleted or their infiltration into the tumor is blocked, tumor-specific Teffs are still able to infiltrate the tumor and mount an anti-tumor cytotoxic response. Although the tumors were not eliminated in this model system, the experiments clearly demonstrated proof-of-principle and mechanism of action. Perhaps a combination therapy with a checkpoint inhibitor or other immunomodulatory treatment would lead to a more complete and durable response. Regardless, these important experiments both advance our understanding of the mechanisms by which tumors evade patient immunity and may impact the development of new treatment strategies for treating cancer patients.
Reference
Chang D, et al., 2016. Anti-CCR4 monoclonal antibody enhances antitumor immunity by modulating tumor-infiltrating Tregs in an ovarian cancer xenograft humanized mouse model.Oncoimmunology. Dec 10;5(3):e1090075.
PMID: 27141347
http://www.ncbi.nlm.nih.gov/pubmed/27141347