Amyotrophic Lateral Sclerosis (ALS), or Lou Gehrig's disease, is a devastating and rapidly fatal neurodegenerative disease that affects motor neurons, leading to loss of voluntary muscle movement and breathing. With few FDA-approved treatments of modest efficacy, there remains a critical need for effective therapies. JAX offers a comprehensive suite of ALS efficacy studies, backed by a portfolio of validated mouse models and hiPSC lines to accelerate translational research.
About 90% of ALS cases have no known cause, while the remaining 10% are hereditary. Mutations in over 20 genes have been associated with familial ALS, with four accounting for the majority of cases: SOD1, TDP-43, FUS, and C9orf72. JAX provides validated models covering each of these key genetic variants:
See all ALS Models at JAX
| Strain Name | Phenotype | Disease Latency | Details |
|---|---|---|---|
SOD1-G93A |
| 2 months |
|
B6 SOD1-G93A |
| 1.5 months |
|
| Strain Name | Phenotype | Disease Latency | Details |
|---|---|---|---|
Prp-TDP43A315T |
| 3-4 months |
|
Prp-TDP-43Q331K line 103 |
| 2-3 months |
|
TAR4 |
|
| |
Δ-NLS |
| 2 months |
|
| Strain Name | Phenotype | Disease Latency | Details |
|---|---|---|---|
Tg(C9orf72_3) line 112 |
| 3 months |
|
In addition to ALS mouse models, The Jackson Laboratory also offers a collection of gene-edited hiPSC lines, carrying mutations associated with neuromuscular diseases, providing complementary tools for translational research. Explore our iPSC catalog.
Partner with JAX Preclinical Services to offload the full complexity of your ALS efficacy studies. JAX brings scientific expertise and expert colony management with the following study endpoints:
Consult with JAX About Running Your ALS Study Today
| Study Design | Detail |
|---|---|
| Timeline | 8+ weeks |
| Groups | 12-16 models per group Mice aged 6/7 weeks at the start of study |
| Baseline |
|
| In Vivo | Daily Dosing for 8 weeks. Onset & progression of disease are monitored via electromyography (EMG) and rotarod at various time points
|
| Post-mortem Analysis |
|
B6SJL-Tg(SOD1*G93A)1Gur/J (002726)
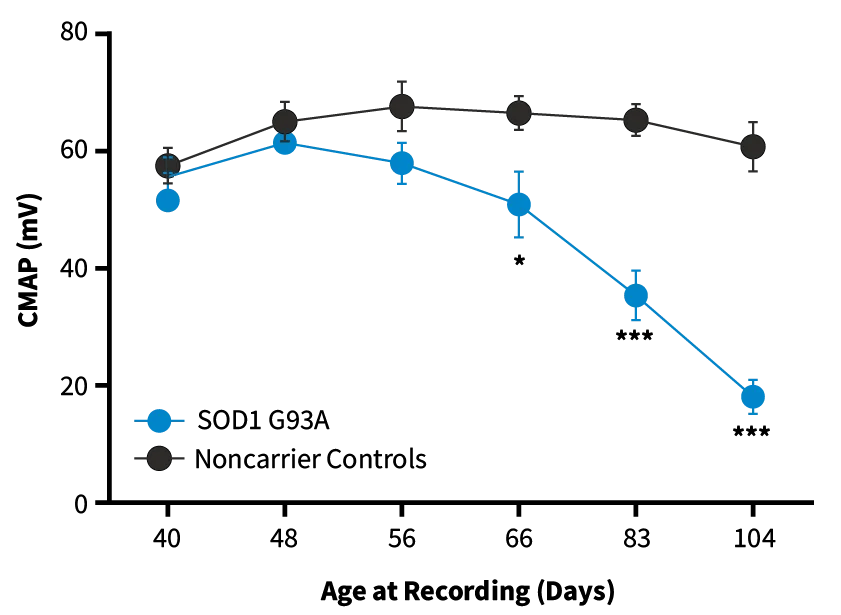
Compound Muscle Action Potential (CMAP). SOD1-G93A mice show decreased CMAP amplitude compared to non-carrier controls as they age, indicating progressive muscle denervation and motor neuron degeneration (* = p<0.05; *** = p<0.001).
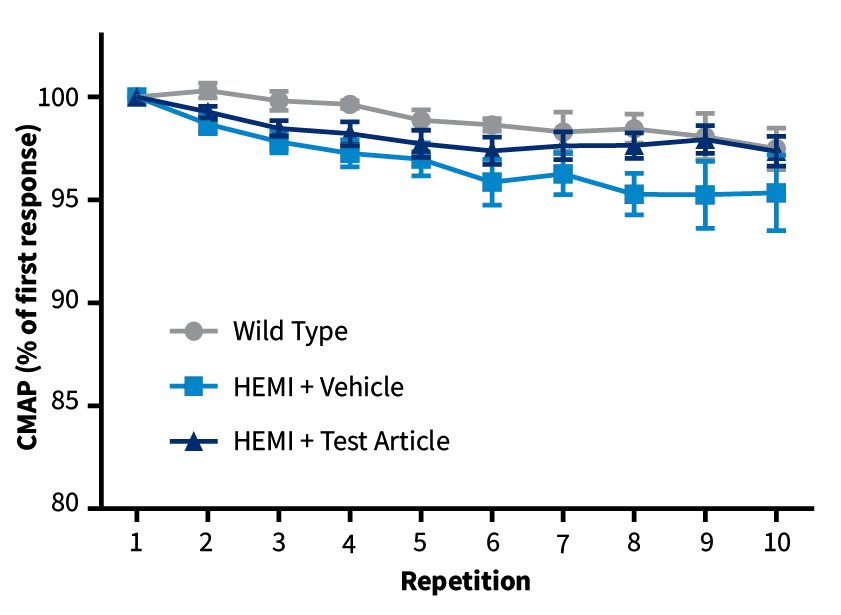
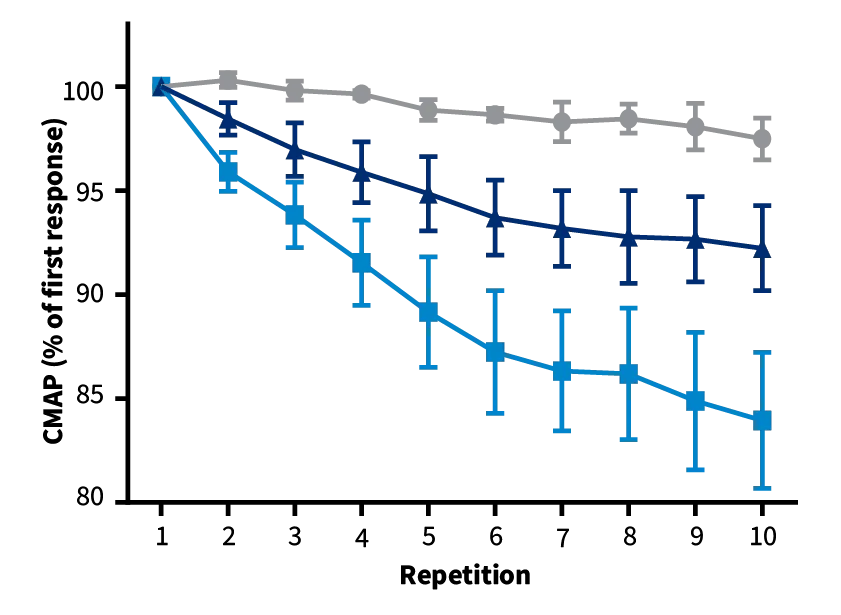
Repetitive Nerve Stimulation (RNS). SOD1-G93A mice (blue and navy) show decreased CMAP values compared to non-carrier controls (grey) during RNS, indicative of reduced neuromuscular junction transmission between muscle fibers and motor neurons. Left graph: prior to disease onset. Right graph: after disease onset, mice treated with the test article show a milder decrement upon RNS, indicative of preserved NMJ transmission.
B6SJL-Tg(SOD1*G93A)1Gur/J (002726)
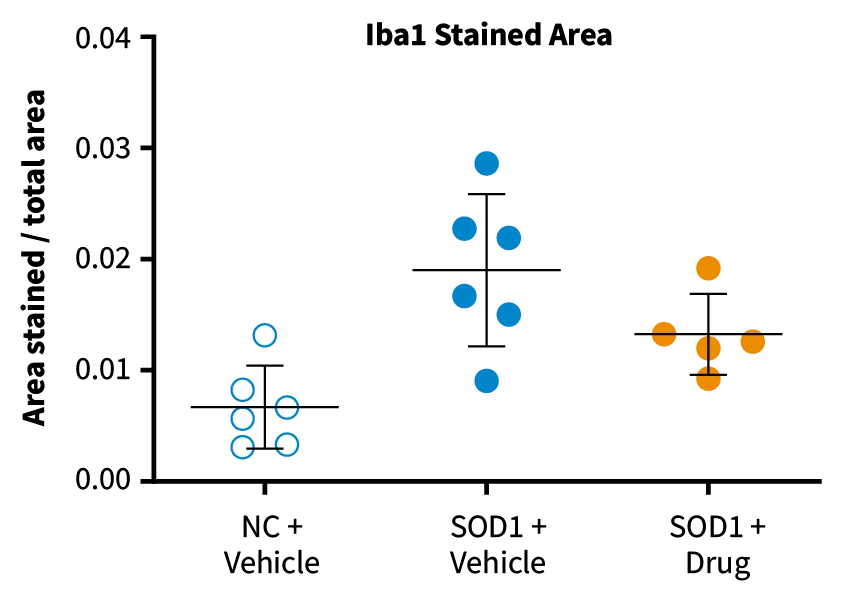
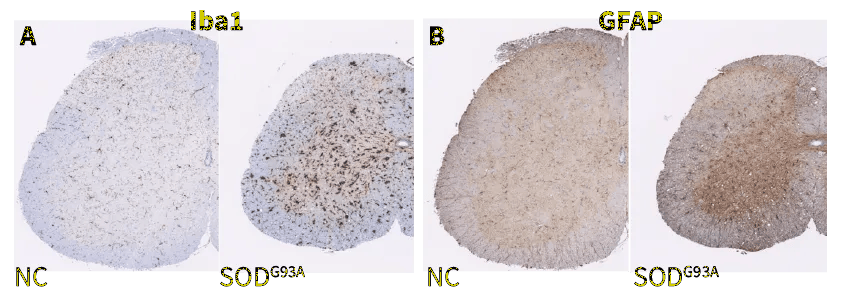
Neuroinflammation, Spinal Cord. SOD1-G93A mice show increased immunoreactivity of microglia (Iba1, A) and astrocytes (GFAP, B) in the spinal cord compared to non-carrier (NC) controls. Drug treatment reduces immunoreactivity in SOD1-G93A mice relative to vehicle-treated SOD1-G93A mice (C).
In this guide, you will find a summary of the best practices and recommendations for designing and conducting preclinical studies using available SOD1-based mouse models of ALS, as well as best practices on breeding and maintaining SOD1 mutant mouse colonies.