Variable patient responses to SARS-CoV-2 infection are mimicked in genetically diverse mice
Research Highlight | July 25, 2023
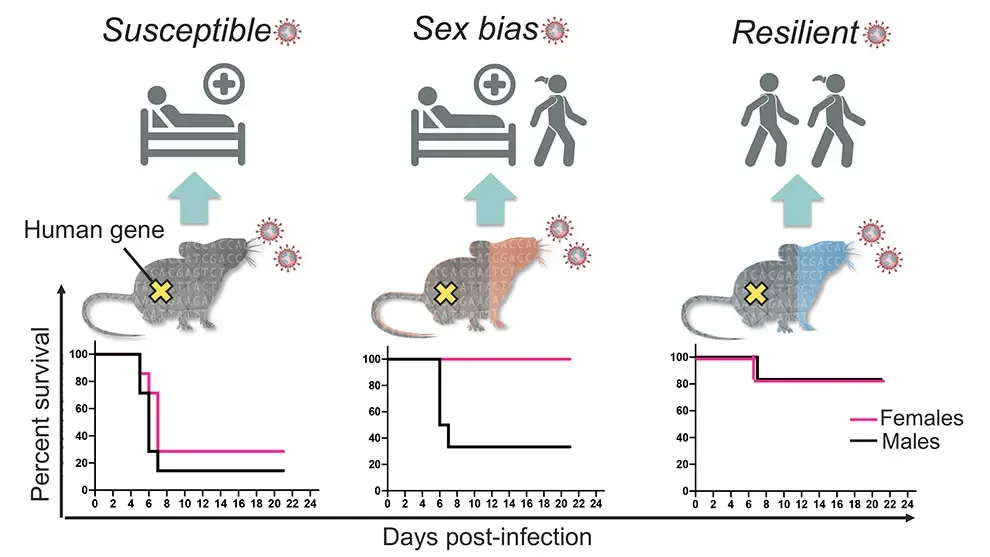

Reproducing variable human response
Early in the COVID-19 pandemic, it became clear that people had hugely variable responses to SARS-CoV-2 infection. Many exhibited no symptoms at all, while a small percentage contracted severe or lethal disease. The timing and strength of innate immune activity and interferon signaling, the front-line cellular defense against microbial infection, were implicated in this variability, but the underlying factors determining disease severity between individuals remained poorly understood.
In response to the pandemic, a mouse model was quickly re-derived that allowed SARS-CoV-2 infection through humanized angiotensin-converting enzyme 2 (hACE2) receptors. At first, the hACE2 was only present in a single inbred mouse line, known as K18-hACE2, which always developed severe/lethal disease. To see whether the variable human response could be replicated in mice, a team of researchers led by The Jackson Laboratory (JAX) Scientific Director and ProfessorNadia Rosenthal, Ph.D., F.Med.Sci., and Rocky Mountain National Laboratories Chief of Innate Immunity and Pathogenesis Sonja Best, Ph.D., crossed the original K18-hACE2 line with other mouse strains that represent broad genetic diversity.
In “Genetically diverse mouse models of SARS-CoV-2 infection reproduce clinical variation in type I interferon and cytokine responses in COVID-19,” apaper published in Nature Communications, the team shows that the resulting F1 (first generation crossed) mice indeed modeled human COVID-19 severity, ranging from asymptomatic to lethal. The original genetic background used for the K18-hACE2 mice (C57Bl/6J) proved to be among the most susceptible, while the F1 crosses from a strain known as PWK were highly resistant to disease. F1 progeny of the other crosses, derived from the inbred strains A/J, 129S1, NOD, NZO, CAST, WSB, BALB/c, and DBA/2, had a range of responses mostly between the two extremes. Interestingly, some of them—CAST, NOD, and WSB—also had sex differences, with consistently different levels of disease severity between F1 males and females.
Developing a preclinical platform
With the mouse panel, the team was able to further investigate differences in the innate immune responses that had been implicated in human patient variability. In particular, type 1 interferon (IFN-1) is essential for control of virus replication, where the timing and regulation of the response plays key roles in determining disease severity. If the response is delayed, viral replication and spread can proceed unchecked during the early stages of infection. At the same time, failure to regulate it and reduce signaling once acute infection is over can lead to ongoing inflammation and adverse health consequences.
The research team found that the highly resistant PWK F1 mice exhibited early control of virus replication in the lungs, with phased amplification and resolution of pro-inflammatory responses and prevention of virus dissemination to other organs. In contrast, F1 crosses with the more susceptible strains exhibited relatively inefficient IFN-1 expression in the lung, failed control of virus replication, and dysregulated pro-inflammatory responses. An outlier was WSB, which had high early IFN-1 expression but also high early virus burden in the lung, and clearance was delayed in a manner similar to mice with low IFN-1 expression. WSB may therefore prove valuable for investigating additional pathological responses associated with high IFN-1 expression but low antiviral activity.
There are many gaps in our current knowledge that remain to be addressed, including the exact mechanisms of innate immune control of virus replication, the events needed for a well-orchestrated inflammatory response, the molecular mechanisms of sex-dependent disease severity, and longer-term implications for tissue repair and lung function. Developing a preclinical platform for linking disease outcome to patient genetic features promises to bridge the knowledge gap in our understanding of the underlying differences in COVID-19 susceptibility, facilitating the development of precise models for rapid diagnoses, mechanistic studies and therapeutic intervention strategies.