
Whole exome and whole genome sequencing have greatly improved the diagnosis of Mendelian disorders and diseases. And it’s getting better, as the diagnostic yield has improved from 16%-25% in early studies to 35%-60% currently, sparing many patients and families from a long and arduous diagnostic odyssey to find the root cause of disease.
Nonetheless, a high proportion of cases still remain unresolved, underscoring the need for better tools for finding disease-causing genetic variants. A recent paper presents a new algorithm, named SQUIRLS, that provides accurate, automated detection of variants that affect mRNA splicing, which underlie many Mendelian disorders.
When a messenger RNA (mRNA) is first transcribed from its DNA code, it carries the information needed to make a functional protein, but not in final form. After it exits the nucleus as a pre-mRNA, and before it’s translated to a protein by a ribosome, it undergoes what is known as the splicing process. Certain segments (introns) are deleted and others (exons) are retained, and the remaining exons are spliced together into a mature mRNA transcript that will yield a protein with the correct amino acid sequence.
Splicing is tightly controlled, but mutations and other variations in the DNA sequence may disrupt it. Defective splicing can lead to skipped exons, included introns and other errors in the mRNA transcript that is translated. The resulting protein is unlikely to function properly, potentially leading to disease. In fact, research has shown that at least 15 percent of all disease-causing genetic variants are ones that affect pre-mRNA splicing. It is therefore vital to identify and interpret variants in the DNA sequences that alter splicing as part of clinical sequencing and diagnostics.
Automating splice variant detection
Certain splice-altering variants are relatively easy to identify, occurring in two-base sequences at either end of introns. These “canonical” AG/GT sequences are highly conserved, meaning that they remain the same across proteins and even between species, and any change to them is a red flag in diagnostic whole genome or whole exome sequence analysis. But there are many other genetic variants at other locations within the genome that affect splicing function. These non-canonical variants are far more difficult to identify and associate with a disease diagnosis.
To address the problem, a team led by Jackson Laboratory (JAX) Professor Peter Robinson, M.D., M.SDevelops algorithms and software for the analysis of exome and genome sequences. Peter Robinson ,M.D., MSc., and Associate Computational Scientist Daniel Danis, Ph.D.I work on development of new algorithms designed to analyze big data to enable precision medicineDaniel Danis , Ph.D., used machine learning and datasets of splice variants associated with Mendelian (single-gene, inherited) disorders to develop a new algorithm to improve the diagnostic process. In “Interpretable prioritization of splice variants in diagnostic next-generation sequencing,” a paper published in the American Journal of Human Genetics, the researchers present “super quick information-content random-forest learning of splice variants,” or SQUIRLS.
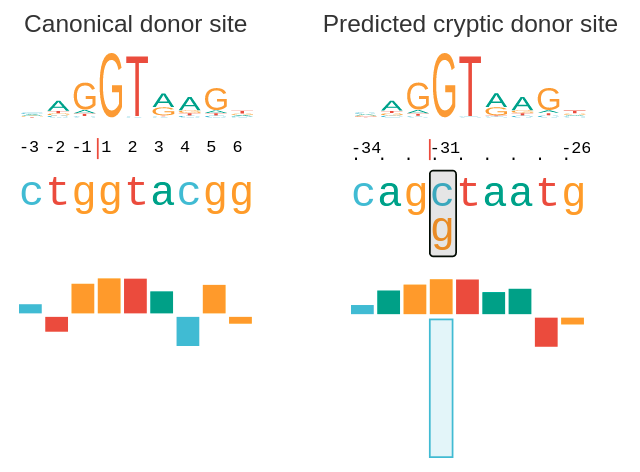
SQUIRLS was trained on a large dataset of non-canonical splice-affecting variants, and as such performs particularly well in identifying difficult-to-classify variants located outside the canonical sequences. Using a test dataset, the team demonstrated that SQUIRLS matches or outperforms four previously published algorithms and methods in assessing and prioritizing splice variants in clinical sequence data. SQUIRLS can be used either on its own or as part of a diagnostic exome/genome pipeline to improve causal splice variant recognition.
Importantly, it also outputs results with visualizations and assessments of each feature, allowing users to quickly see how SQUIRLS prioritized the variants, a necessity for busy clinical workplaces.
Genomic medicine
Initiatives such as the UK 100,000 Genomes project are making genomic medicine part of everyday healthcare and expanding the use of genome data in rare disease diagnosis. It is therefore crucial to continue to improve diagnostic yields while maximizing speed, efficiency, and ease of use. SQUIRLS represents an important step forward for the Mendelian disorder diagnostic toolkit, providing fast, accurate analysis of splice-affecting variants that were previously difficult to assess.