Uncovering the mechanism(s) of ALS
Understanding the biological mechanism(s) leading to motor neuron degeneration in amyotrophic lateral sclerosis (ALS or Lou Gehrig's Disease) is key to the development of new therapies for this devastating disease. A new publication in Science Translational Medicine (de Boer, A. S. et al. 2014) provides genetic validation that microglia from ALS mice (B6SJL-Tg(SOD1*G93A)1Gur/J 002726, commonly called SOD1G93A mice) are toxic to normal human motor neurons. The researchers also found that blockage of signaling through the prostaglandin D receptor (Ptgdr, or DP1), using either a small-molecule antagonist or through genetic deletion of the receptor, could restore motor neuron survival. SOD1G93A mice genetically null for Ptgdr showed increased life span, providing hope this pathway may be a new therapeutic target for ALS patients.
In Vitro Analysis of Motor Neuron Toxicity. Human embryonic stem cells were differentiated into motor neurons and isolated by cell sorting based on expression of GFP driven by the motor neuron specific promoter Hb9. Motor neurons co-cultured with glia (astrocytes and microglia) from SOD1G93A mice showed diminished survival. de Boer, A. S. et al. found that blocking signaling through the prostaglandin 1 receptor in microglia diminished toxicity and improved motor neuron survival.
Delineating the receptor leading to motor neuron toxicity
Prior research using an in vitro co-culture approach provided evidence that astrocytes and microglia expressing a human mutation in superoxide dismutase 1 (SOD1G93A) produce a toxic substance that causes motor neuron degeneration.
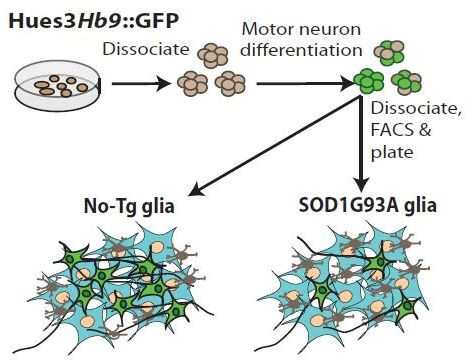
Also, when normal glia were treated with prostaglandin D2, similar results were observed. The new publication by de Boer, A. S. et al. further defined this mechanism. Prostaglandin D2 acts through two different receptors, DP1 and DP2. Determination of the receptor pathway responsible was accomplished using small-molecule antagonists in the co-culture system. An antagonist that binds both receptors modestly diminished the toxicity to motor neurons from SOD1G93A derived glia. Another antagonist specific for DP1 alone eliminated the toxicity of these cells. When co-cultures of motor neurons and normal glia were treated with a DP1 specific agonist, motor neuron toxicity was observed implicating DP1 as the responsible receptor pathway. The researchers also demonstrated that it was DP1 on glia, not motor neurons, that prevented cell death by pre-treating glia with these compounds prior to co-culture with the motor neurons and obtaining similar results. Together, these experiments demonstrated glial-specific signaling through DP1 was responsible for induced death of motor neurons.
Genetic ablation of DP1 improves survival
An important remaining question was whether blocking of DP1 signaling in vivo also improves motor neuron survival. This question was examined by crossing the SOD1G93A mice with another strain containing a null mutation in DP1 (Ptgdrtm1Sna). The first step in this evaluation was to validate the impact of the DP1 mutation at the cellular level by performing the in vitro co-culture experiments with glia from the double mutant mice. When SOD1G93A DP1 +/- or DP1-/- glia were cultured with motor neurons, percent survival of the motor neurons was similar to that observed when using the DP1 antagonist (improved survival). Also, when transgene negative DP1 +/+, DP1 +/- or DP1-/- glia were treated with the agonist, only the DP1 wild type and heterozygous glia showed induced motor neuron toxicity. These experiments demonstrated that glia from SOD1G93A mice genetically ablated of the DP1 receptor respond in the same manner as cells treated with the small-molecule antagonist, further validating the importance of the DP1 pathway in ALS.
For in vivo analysis, SOD1G93A mice that were DP1 +/+, DP1 +/- or DP1-/- were aged to measure differences in long-term survival. Unfortunately, disease onset was not eliminated and occurred at 108 ± 2 days and 111 ± 3 days for SOD1G93A DP1 +/- and SOD1G93A DP1 -/- mice, respectively. However, survival was extended in both SOD1G93A DP1 +/- and DP1-/- mice, with an overall 6.7% increase in total lifespan. Spinal cords were collected from mice at day 100 and stained with anti-ChAT antibody to quantitate motor neurons. SOD1G93A DP1 +/- and DP1-/- mice had increased motor neuron survival compared to SOD1G93A DP1 +/+ mice. In addition, SOD1G93A mice with deletions in DP1 had fewer activated microglia in the spinal cord as compared to SOD1G93A DP1 +/+ mice.
These results show that blocking the DP1 receptor does not eliminate development of ALS in the mice but does diminish microglia activation, improve survival of motor neurons, and extend lifespan. Uncovering and validating this mechanism of disease in ALS is an important new development that will hopefully translate to clinical application.